The Biology of Aging: Chemistry Behind Growing Old
Aging is among the most universal of all biological phenomena, yet it remains one of the least completely understood. Every organism equipped with a soma, from the microscopic nematode Caenorhabditis elegans to the long-lived bowhead whale, undergoes progressive physiological deterioration over time. What drives this deterioration is not a single molecular event but an interlocking network of chemical and cellular changes that accumulate across a lifetime. These changes alter gene expression, damage macromolecules, remodel tissue architecture, and ultimately erode the homeostatic mechanisms that keep organisms functional. Understanding the chemistry of aging requires moving across multiple scales simultaneously: from the atomic chemistry of reactive oxygen species, to the molecular biology of telomeres and epigenetic marks, to the cell biology of senescence, to the systems-level consequences for tissue and organ function.
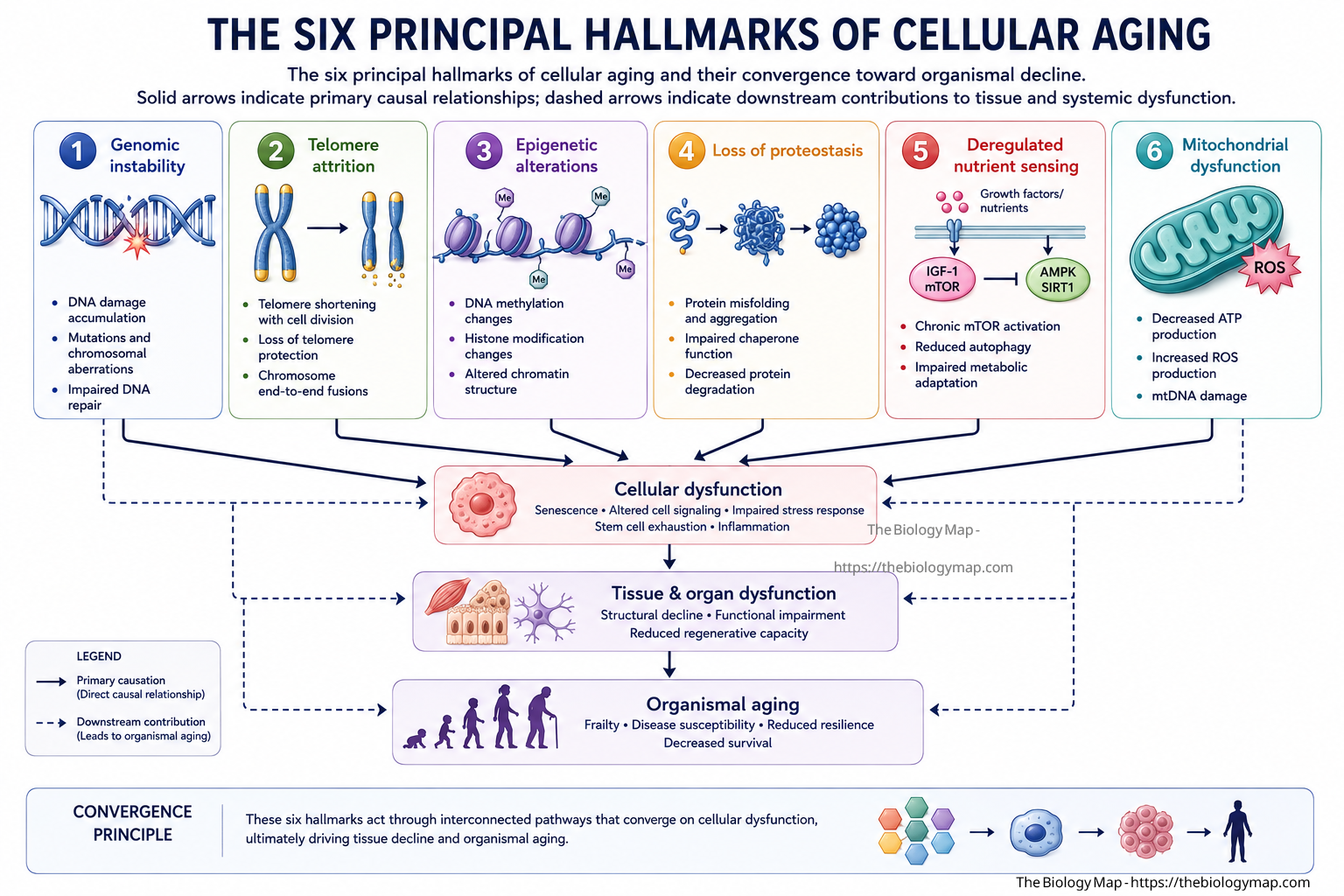
Figure 1: The six principal hallmarks of cellular aging and their convergence toward organismal decline. Solid arrows indicate primary causal relationships; dashed arrows indicate downstream contributions to tissue and systemic dysfunction.
The scientific study of aging, broadly termed geroscience, has accelerated dramatically over the past three decades. The identification of conserved longevity pathways, the cataloguing of molecular hallmarks, and the development of interventions that extend lifespan in model organisms have transformed aging from a topic of philosophical contemplation into a tractable area of molecular biology. As of 2026, the field is navigating a particularly productive period: large-scale longitudinal omics studies are beginning to resolve the temporal ordering of aging processes in humans, and the first senolytic compounds are moving through clinical trials. This article examines the chemical and molecular foundations of aging in depth, with particular attention to the mechanisms that have proven most amenable to experimental interrogation and those most likely to yield therapeutic insights.
The Molecular Basis of Biological Time: Telomeres and Replicative Senescence
At the physical ends of every linear eukaryotic chromosome sit telomeres, repetitive hexanucleotide sequences (TTAGGG in humans) bound by a specialised protein complex known as shelterin. Telomeres serve as protective caps that prevent chromosome ends from being recognised as double-strand breaks and subjected to inappropriate repair or degradation. Their chemistry is directly linked to the passage of cellular time because of a fundamental limitation in DNA replication known as the end-replication problem.
DNA polymerases require a free 3′-hydroxyl group on a primer and can only synthesise in the 5′-to-3′ direction. As a consequence, the lagging strand template cannot be fully copied at its very terminus after the RNA primer for the most distal Okazaki fragment is removed. Each round of cell division therefore results in a net loss of approximately 50 to 200 base pairs of telomeric sequence. After enough divisions, telomeres reach a critically short length, at which point shelterin can no longer maintain the protective T-loop structure. The exposed chromosome end activates a DNA damage response, predominantly mediated by ataxia-telangiectasia mutated (ATM) kinase and ataxia telangiectasia and Rad3-related (ATR) kinase, which phosphorylate histone H2AX and recruit downstream effectors including p53 and the cyclin-dependent kinase inhibitor p21. The cell arrests in G1 phase and enters a state of permanent proliferative arrest: replicative senescence, first characterised by Leonard Hayflick in the early 1960s.
Telomerase and Its Limitations
The enzyme telomerase counteracts telomere shortening by extending the 3′ overhang using an internal RNA template (TERC) and a reverse transcriptase catalytic subunit (TERT). In humans, telomerase is robustly active in germline cells, ensuring transgenerational telomere integrity, and is transiently expressed in progenitor and stem cell populations. However, the vast majority of somatic cells either lack telomerase expression or express it at levels insufficient to fully compensate for replication-associated attrition. This intentional constraint is thought to have evolved partly as a tumour-suppressor mechanism: limiting the proliferative lifespan of individual cells reduces the probability that a given cell accumulates the mutational burden required for malignant transformation.
The tradeoff is that tissues relying on stem cell pools for self-renewal, including the intestinal epithelium, the haematopoietic system, and the epidermis, become progressively compromised as stem cell telomeres shorten over decades. Studies in mice with engineered telomerase deficiency show accelerated aging phenotypes across multiple organs, and in humans, mutations in TERT or TERC cause the premature aging syndrome dyskeratosis congenita. Conversely, long telomeres and high telomerase activity in long-lived species such as the bowhead whale have raised interest in telomere biology as a determinant of maximum lifespan at the species level.
Genomic Instability and the Accumulating Mutational Burden
Beyond the predictable attrition of telomeric repeats, the genome is subject to continuous chemical assault that introduces lesions throughout its roughly 3.2 billion base pairs. These lesions arise from both endogenous and exogenous sources, and their incomplete or inaccurate repair drives the accumulation of somatic mutations that characterises aging tissue.
Endogenous sources include the spontaneous hydrolysis of the N-glycosidic bond linking nucleobases to deoxyribose, producing abasic (apurinic or apyrimidinic) sites at an estimated rate of approximately 10,000 events per cell per day. Deamination of cytosine to uracil occurs at a rate of several hundred events per cell per day and, if unrepaired before replication, produces C-to-T transition mutations. Reactive oxygen species generated as byproducts of oxidative metabolism produce a wide spectrum of oxidative base modifications, the most studied of which is 8-oxo-7,8-dihydroguanine (8-oxoG), which mispairs with adenine during replication to produce G-to-T transversions.
DNA Repair Capacity and Its Decline
Cells are equipped with an elaborate suite of repair pathways calibrated to different lesion types. Base excision repair (BER) handles small, non-helix-distorting lesions such as 8-oxoG and uracil. Nucleotide excision repair (NER) removes bulky adducts and photoproducts. Mismatch repair (MMR) corrects replication errors, and homologous recombination (HR) and non-homologous end joining (NHEJ) address double-strand breaks. A consistent observation across multiple model systems is that the efficiency and fidelity of these repair pathways declines with age. The expression of key repair factors including BRCA1, RAD51, and components of the MRN complex (MRE11-RAD50-NBS1) decreases in aged tissues. NHEJ, the predominant double-strand break repair pathway in post-mitotic cells, becomes increasingly error-prone, contributing to structural chromosomal rearrangements.
The consequence is a somatic mutational burden that accumulates at an estimated rate of approximately 1 to 2 mutations per billion base pairs per cell per year in most normal human tissues, though this rate varies substantially by tissue and by exposure history. Deep sequencing studies of normal human tissues, including skin, oesophagus, and haematopoietic cells, have revealed that by middle age, many apparently normal tissue regions already harbour driver mutations in canonical cancer genes. This observation has reframed the relationship between aging and cancer: oncogenesis is not merely a consequence of single catastrophic events but of a gradual expansion of clonal populations carrying fitness-enhancing mutations within an aging tissue microenvironment.
Epigenetic Reprogramming and the Aging Epigenome
If the genome encodes what a cell can do, the epigenome encodes what it currently does. Epigenetic modifications regulate chromatin accessibility and gene expression without altering the underlying nucleotide sequence, and they are profoundly reshaped during aging in ways that alter cellular identity, stress responses, and tissue function.
DNA methylation at cytosines within CpG dinucleotides is the most intensively studied epigenetic mark in the context of aging. Two opposing trends characterise the aging methylome. Globally, there is a generalised loss of DNA methylation, particularly at repetitive elements such as LINE-1 retrotransposons and satellite sequences, which are normally maintained in a hypermethylated and transcriptionally silenced state. When this silencing is lost, retrotransposon transcription and even mobilisation can resume, introducing insertional mutations and triggering innate immune signalling via cytosolic nucleic acid sensors. Simultaneously, specific CpG islands in gene promoters become aberrantly hypermethylated with age, silencing genes involved in stress response, differentiation, and tumour suppression.
Epigenetic Clocks and Biological Age
Steve Horvath’s landmark 2013 work demonstrated that DNA methylation at a curated set of CpG sites predicts chronological age with remarkable accuracy across diverse tissues and cell types. The resulting “epigenetic clock” and its successors (GrimAge, PhenoAge, DunedinPACE) measure not merely elapsed time but the rate of biological aging. Individuals whose epigenetic age exceeds their chronological age by several years show elevated risks of age-related disease and all-cause mortality independent of conventional clinical risk factors. These clocks have also become indispensable research tools: interventions that slow or reverse the epigenetic clock in animal models are now considered strong candidates for further translational development.
Histone modifications form the second major layer of epigenetic regulation disrupted during aging. The overall abundance of histones declines with age as histone gene expression is attenuated, and the distribution of activating marks (H3K4me3, H3K36me3) and repressive marks (H3K9me3, H3K27me3) shifts in ways that derepressed previously silenced genomic regions while silencing others. Heterochromatin, the dense, transcriptionally inactive form of chromatin concentrated at pericentromeric and telomeric regions, is progressively lost during aging in a process termed heterochromatin erosion. This erosion contributes to transcriptional noise, the increase in cell-to-cell variability in gene expression that is a hallmark of aged tissue, and which may be particularly disruptive to processes requiring precise threshold responses such as immune activation or stem cell fate decisions.
Reactive Oxygen Species and the Oxidative Chemistry of Cellular Damage
The free radical theory of aging, proposed by Denham Harman in 1956 and extended into the mitochondrial free radical theory by the same author in 1972, posited that reactive oxygen species (ROS) generated during aerobic metabolism are the primary molecular drivers of aging. While subsequent decades of research have substantially refined this view, recognising that ROS also serve essential signalling functions and that antioxidant supplementation does not uniformly extend lifespan, oxidative chemistry remains central to understanding the molecular damage that accumulates during aging.
The electron transport chain embedded in the inner mitochondrial membrane couples the sequential oxidation of NADH and FADH2 to the pumping of protons across the membrane, generating the electrochemical gradient that drives ATP synthase. At complexes I and III, a small fraction of electrons, estimated at 0.1 to 2 percent under normal physiological conditions, escape the chain and reduce molecular oxygen to superoxide (O2 radical anion). Superoxide is rapidly dismutated to hydrogen peroxide (H2O2) by manganese superoxide dismutase (MnSOD) in the mitochondrial matrix or copper-zinc superoxide dismutase (Cu/ZnSOD) in the cytoplasm. H2O2 is itself a relatively mild oxidant but serves as the substrate for the Fenton reaction: in the presence of ferrous iron (Fe2+), H2O2 is reduced to the hydroxyl radical (OH radical), arguably the most reactive species in cellular chemistry, with a half-life on the order of nanoseconds and reactivity sufficient to oxidise any adjacent macromolecule without discrimination.
Mitochondrial Dysfunction as a Driver of Aging
With age, mitochondrial function deteriorates along multiple dimensions. Mutations accumulate in mitochondrial DNA (mtDNA), which lacks the protective histone coating of nuclear DNA and is in close proximity to the electron transport chain and its associated ROS production. Large-scale mtDNA deletions accumulate in post-mitotic tissues including neurons and cardiomyocytes, reaching levels in individual cells that impair oxidative phosphorylation. Mitochondrial dynamics, the coordinated cycle of fusion and fission that maintains network quality, and mitophagy, the selective autophagic clearance of damaged mitochondria, both become less efficient with age, allowing dysfunctional organelles to persist and amplify ROS production.
The NAD+ metabolite pool, essential for both mitochondrial redox reactions and as a substrate for the sirtuin family of NAD+-dependent deacylases, declines substantially with age in multiple tissues. Sirtuins (SIRT1 through SIRT7 in humans) regulate a broad array of targets relevant to aging, including the master mitochondrial biogenesis regulator PGC-1 alpha, the stress response transcription factor FOXO3, and histones. The decline in NAD+ therefore cascades into reduced sirtuin activity, impaired mitochondrial biogenesis, and attenuated stress resistance. Supplementation with NAD+ precursors such as nicotinamide riboside (NR) or nicotinamide mononucleotide (NMN) restores NAD+ levels in aged rodent tissues and improves a range of aging-associated phenotypes, and early human trials have confirmed the bioavailability and safety of these compounds, though functional endpoints in humans remain under active investigation as of 2026.
Proteostasis and the Chemistry of Protein Maintenance
Every functional protein must fold correctly, localise appropriately, interact with the right partners, and be degraded at the right time. The ensemble of molecular machinery that maintains this state is termed the proteostasis network, and its progressive failure is a defining feature of cellular aging with direct mechanistic links to a large class of age-associated diseases.
Newly synthesised polypeptides emerging from the ribosome are at risk of misfolding, particularly under cellular stress. Molecular chaperones of the heat shock protein (HSP) family, including HSP70, HSP90, and the chaperonin complex GroEL/GroES (or TRiC/CCT in eukaryotes), recognise exposed hydrophobic stretches on unfolded or partially folded clients and facilitate correct folding in an ATP-dependent manner. With aging, the inducibility of the heat shock response declines due to reduced activity of the master transcription factor HSF1, leaving aged cells with a diminished capacity to upregulate chaperone expression under stress. Baseline chaperone levels also decline in many aged tissues.
Proteasomal Degradation and Autophagy in Aging
Misfolded and damaged proteins that cannot be refolded are targeted for degradation, primarily through the ubiquitin-proteasome system (UPS) or through autophagy. In the UPS, substrates are covalently tagged with chains of the 76-amino-acid protein ubiquitin by a three-enzyme cascade (E1 activating, E2 conjugating, E3 ligating enzymes), and the tagged substrate is unfolded and threaded through the 20S proteolytic core of the 26S proteasome. Both the activity of individual proteasomal subunits and the concentration of intact 26S complexes decline measurably in aged cells and tissues. The accumulation of oxidatively modified proteins, which may cross-link and resist proteasomal processing, contributes to this impairment by partially blocking the barrel of the proteasome.
Macroautophagy, the sequestration of cytoplasmic material including organelles and protein aggregates in double-membrane autophagosomes that subsequently fuse with lysosomes, is an essential complement to the UPS for clearing bulkier substrates. Chaperone-mediated autophagy (CMA), the selective import of substrate proteins directly into the lysosomal lumen via the LAMP2A receptor, handles a distinct substrate pool. Both pathways show documented age-related decline. Lysosomal acidity decreases with age, reducing the activity of acid hydrolases. LAMP2A levels at the lysosomal membrane decline. The consequence of these converging failures is the accumulation of insoluble protein aggregates within aged cells, a pathological feature most prominently manifested in neurodegenerative diseases such as Alzheimer’s disease (amyloid-beta and tau aggregation), Parkinson’s disease (alpha-synuclein aggregation), and Huntington’s disease (polyglutamine-expanded huntingtin).
Cellular Senescence and the Senescence-Associated Secretory Phenotype
Cellular senescence is a state of stable, essentially irreversible cell cycle arrest accompanied by profound changes in cell morphology, metabolism, and secretory behaviour. Originally described as the endpoint of replicative exhaustion in culture, it is now recognised as a physiologically programmed response to multiple forms of cellular damage and stress, including oncogene activation, oxidative damage, and paracrine signals from neighbouring senescent cells.
The molecular execution of senescence converges on two principal pathways. The p53-p21 axis is engaged primarily in response to DNA damage and short telomeres: p53, stabilised by ATM/ATR-mediated phosphorylation, transcriptionally activates the cyclin-dependent kinase inhibitor CDKN1A (encoding p21), which inhibits CDK2 and blocks Rb phosphorylation, maintaining Rb in its growth-suppressive, E2F-sequestering form. The p16INK4a-Rb axis is more often activated by oncogene-induced senescence and persistent epigenetic stress: p16, encoded by the CDKN2A locus, directly inhibits CDK4 and CDK6, preventing Rb phosphorylation by a distinct upstream route. In deep senescence, both pathways are typically co-activated, producing a self-reinforcing arrest that is highly resistant to mitogenic signals.
The SASP and Its Systemic Consequences
What distinguishes senescent cells as actors in tissue aging rather than mere bystanders is the senescence-associated secretory phenotype (SASP), a complex and context-dependent secretome comprising proinflammatory cytokines (IL-6, IL-1 alpha, IL-8/CXCL8), matrix metalloproteinases (MMP-1, MMP-3, MMP-9), growth factors (amphiregulin, HGF), and bioactive lipid mediators. The SASP is regulated at the transcriptional level largely by NF-kappaB and C/EBPbeta, which are activated downstream of cytoplasmic DNA sensing via the cGAS-STING pathway. Cytoplasmic DNA accumulates in senescent cells from multiple sources: cytoplasmic chromatin fragments (CCFs) derived from nuclear envelope rupture, cytosolic mtDNA released from compromised mitochondria, and transcripts of reactivated retrotransposons.
The SASP exerts pleiotropic effects on the tissue microenvironment. In acute physiological contexts, including wound healing, embryonic development, and anti-tumour immune surveillance, the SASP is beneficial: it recruits immune cells, promotes tissue remodelling, and signals the clearance of potentially malignant senescent cells. In the context of chronic aging, however, the failure of immune-mediated clearance, partly because the immune system itself ages and loses effector function, allows senescent cells to accumulate and maintain persistent SASP signalling. This creates a state of inflammaging, the low-grade, sterile, chronic inflammation that characterises aged organisms and that correlates with risk of virtually all major age-related diseases including cardiovascular disease, type 2 diabetes, neurodegeneration, and cancer.
The therapeutic potential of targeting senescent cells has been substantiated by multiple animal studies showing that genetic or pharmacological elimination of senescent cells (using navitoclax, dasatinib plus quercetin, or the ABT-263 inhibitor class) extends median and maximal lifespan, delays the onset of multiple age-related pathologies, and improves physical function in already-aged animals. The first human trials of senolytic compounds in conditions including idiopathic pulmonary fibrosis, diabetic kidney disease, and frailty are ongoing as of 2026, with early data suggesting tolerability and, in some cases, reductions in circulating SASP markers.
Nutrient Sensing Pathways and the Biochemistry of Longevity Regulation
A convergent finding from decades of genetic and pharmacological studies in organisms ranging from yeast to primates is that the rate of aging is not fixed but can be substantially modulated by interventions targeting nutrient-sensing signalling networks. This indicates that longevity is regulated, at least in part, by biochemical pathways that evolved to match anabolic growth and reproductive activity to nutrient availability.
The insulin and insulin-like growth factor 1 (IGF-1) signalling (IIS) pathway is the most evolutionarily ancient and most extensively characterised longevity pathway. Activation of the insulin receptor or IGF-1 receptor (IGF1R) triggers sequential activation of IRS-1, PI3K, PDK1, and AKT, which phosphorylates and excludes the FOXO family of transcription factors from the nucleus. FOXOs regulate a broad transcriptional programme encompassing stress resistance genes, autophagy regulators, and proteostasis components. Reduction in IIS signalling, whether by mutation of the insulin receptor homologue daf-2 in C. elegans, mutations in the IGF-1 receptor or growth hormone receptor in mice, or naturally occurring low-IGF-1 states in certain human populations such as individuals with Laron syndrome, consistently extends lifespan and healthspan. In daf-2 mutant nematodes, lifespan extension exceeds 100 percent and requires the FOXO homologue DAF-16, establishing that longevity depends on FOXO-mediated transcriptional reprogramming rather than simply reduced growth.
mTOR, AMPK, and the Sensing of Energetic State
The mechanistic target of rapamycin complex 1 (mTORC1) integrates signals from amino acid availability (detected via the Ragulator-Rag GTPase system at the lysosomal surface), energy status (AMPK-mediated phosphorylation of Raptor and TSC2), and growth factor signalling (AKT-mediated TSC2 inhibition) to set the rate of protein synthesis, ribosome biogenesis, and anabolic metabolism broadly. Chronic mTORC1 activation promotes growth but suppresses autophagy, attenuates stress responses, and accelerates several aging phenotypes. Pharmacological inhibition of mTORC1 with the allosteric inhibitor rapamycin extends lifespan in mice even when administration begins in mid-life, a finding of considerable translational relevance. The mechanism involves multiple effectors: re-activation of autophagy, attenuation of translation of specific proinflammatory mRNAs via effects on the 4E-BP1/eIF4E axis, and reduced cellular senescence entry.
AMP-activated protein kinase (AMPK), activated when cellular ATP is depleted and AMP/ADP ratios rise, acts in many respects as a molecular counterpart to mTORC1: it phosphorylates and inhibits the mTORC1 activator Raptor, activates ULK1 to initiate autophagy, stimulates mitochondrial biogenesis via PGC-1 alpha, and promotes catabolic metabolism. Caloric restriction (CR), the most reproducible non-genetic intervention for lifespan extension across species, activates AMPK and simultaneously reduces IIS and mTORC1 signalling. The dietary restriction mimetics metformin (which activates AMPK partly through inhibition of mitochondrial complex I) and rapamycin are therefore mechanistically rational candidates for geroscience intervention, and both are under investigation in large clinical trials, including the TAME (Targeting Aging with Metformin) trial, which aims to demonstrate that metformin can delay the onset of a cluster of age-related conditions in humans.
Inflammaging: Chronic Inflammation as a Molecular Feature of Aging
The term inflammaging, coined by Claudio Franceschi and colleagues in the early 2000s, describes the chronic, low-grade, systemic inflammation that is an invariant feature of mammalian aging and a major driver of age-related disease. Unlike acute inflammation, which is self-limiting and resolves once the inciting stimulus is cleared, inflammaging is persistent and is driven by the accumulation of multiple chronic stimuli that individually activate innate immune pathways.
Major contributors include the SASP from accumulating senescent cells; the activation of the cGAS-STING pathway by cytoplasmic DNA from mitochondria, retrotransposons, and nuclear envelope rupture; the translocation of gut microbiota-derived lipopolysaccharide (LPS) and other pathogen-associated molecular patterns (PAMPs) across an aging and increasingly permeable intestinal epithelium; the declining clearance of apoptotic cell debris (efferocytosis) as macrophage function deteriorates; and the accumulation of oxidised lipid species and advanced glycation endproducts (AGEs) that act as damage-associated molecular patterns (DAMPs) recognised by pattern recognition receptors including Toll-like receptors (TLRs) and the NLRP3 inflammasome.
The NLRP3 inflammasome, a multiprotein complex that processes pro-IL-1 beta and pro-IL-18 into their mature, secreted forms via caspase-1, is of particular interest because its activation is driven by multiple aging-relevant signals including ATP, uric acid crystals, mitochondrial ROS, and cholesterol crystals. NLRP3 inhibitors including MCC950 and its successors have shown efficacy in reducing inflammation and improving outcomes in aged animal models, and clinical trials in conditions including heart failure and atherosclerosis are underway.
The systemic consequences of inflammaging extend beyond the canonical inflammatory pathologies to include sarcopenia (partially driven by TNF-alpha and IL-6 mediated suppression of myofibrillar protein synthesis and promotion of proteolytic pathways in muscle), insulin resistance (promoted by IKK-beta/NF-kappaB-mediated serine phosphorylation of IRS-1), neurodegeneration (promoted by microglial activation and neuroinflammation), and cancer (through tumour-promoting effects of cytokines on the tissue microenvironment). Addressing inflammaging is therefore increasingly recognised as a potential means of simultaneously reducing risk across the spectrum of age-related disease rather than targeting individual conditions in isolation.
Advanced Glycation Endproducts and the Chemistry of Long-Lived Molecules
Some of the most abundant and chemically stable macromolecules in the body, including collagen, elastin, lens crystallin, and myelin, are not routinely turned over and therefore accumulate non-enzymatic chemical modifications over decades. Chief among these are advanced glycation endproducts (AGEs), the stable, heterogeneous products of the Maillard reaction between reducing sugars and the primary amine groups of lysine residues and the N-terminal amino groups of proteins.
The reaction proceeds through an initial reversible condensation between the carbonyl group of glucose (or other reducing sugars including ribose, fructose, and glyceraldehyde) and a free amine to form a Schiff base, which rearranges to the more stable Amadori product (exemplified by haemoglobin A1c, HbA1c, used clinically to monitor long-term glycaemic control). Amadori products can undergo further oxidation, dehydration, and rearrangement through a complex network of reactions involving reactive 1,2-dicarbonyl intermediates such as methylglyoxal and glyoxal, ultimately yielding irreversible crosslinks and fluorescent products. The most studied human AGEs include carboxymethyllysine (CML), carboxyethyllysine (CEL), and pentosidine. In long-lived proteins such as collagen, AGE content increases essentially linearly with age and substantially faster in individuals with diabetes or chronic hyperglycaemia.
AGEs exert their biological effects through two mechanisms. First, the formation of covalent crosslinks within and between collagen fibrils reduces the elasticity of connective tissues throughout the body, contributing to arterial stiffening (a major cardiovascular risk factor independent of blood pressure), reduced pulmonary compliance, increased lens rigidity culminating in presbyopia, and decreased joint flexibility. Second, AGEs are recognised by the receptor for AGEs (RAGE), a pattern recognition receptor of the immunoglobulin superfamily that upon engagement activates NF-kappaB, amplifying proinflammatory signalling and contributing to the inflammaging milieu. AGE-RAGE signalling is mechanistically linked to diabetic complications, atherosclerosis, and neurodegeneration.
Stem Cell Exhaustion and the Declining Regenerative Capacity of Aging Tissues
Tissue homeostasis in adult organs is maintained by populations of resident stem and progenitor cells that divide to replenish short-lived differentiated cell types lost through normal turnover and injury. The progressive exhaustion of stem cell populations is a late-stage consequence of the upstream molecular events described above, but it constitutes a hallmark in its own right because it directly determines the functional reserve and regenerative capacity of aging tissues.
In the haematopoietic system, haematopoietic stem cells (HSCs) from aged mice and humans show well-characterised functional changes: reduced self-renewal capacity per cell in transplantation assays, skewed differentiation toward myeloid lineages at the expense of lymphoid progenitors (contributing to immunosenescence and the relative lymphopenia of old age), and impaired homing to bone marrow niches. These changes reflect accumulated DNA damage, epigenetic drift, and alterations in the bone marrow microenvironment including reduced production of niche factors such as CXCL12 and stem cell factor by stromal cells. Clonal haematopoiesis of indeterminate potential (CHIP), the clonal expansion of HSCs carrying somatic mutations in genes including DNMT3A, TET2, and ASXL1, affects more than 10 percent of individuals over 70 years of age and confers elevated risk of haematological malignancy and, unexpectedly, cardiovascular disease, the latter attributed to proinflammatory effects of mutant macrophages in atherosclerotic plaques.
Muscle satellite cells, the resident stem cells of skeletal muscle, decline in number and function with age in a manner that contributes directly to sarcopenia. Neural stem cells in the dentate gyrus of the hippocampus and the subventricular zone show reduced proliferative activity in aged rodents and humans, with implications for hippocampal neurogenesis and cognitive function. Intestinal stem cells, despite being among the most rapidly cycling cells in the body, show age-related changes in Wnt signalling responsiveness and Paneth cell niche function that impair regeneration after injury. A common theme across stem cell compartments is that many functional deficits are partially reversible by systemic interventions, such as caloric restriction, heterochronic parabiosis exposing aged animals to young blood, or pharmacological targeting of specific inhibitory signals, indicating that stem cell exhaustion reflects a partially plastic response to a changed systemic environment rather than irreversible intrinsic deterioration.
Intercellular Communication and the Systemic Propagation of Aging
Aging is not merely a cell-autonomous process. Cells communicate continuously with their neighbours and with distant tissues through paracrine and endocrine signalling, and both the content and quantity of these signals change profoundly with age. The study of intercellular communication in aging has been energised by parabiosis experiments beginning in the 1950s and revived with molecular resolution in the 2010s, which demonstrated that exposing aged mice to young circulation through surgical blood-sharing connections could rejuvenate muscle, brain, and liver function.
Subsequent work identified specific factors in young plasma responsible for some of these effects, including growth differentiation factor 11 (GDF11), which showed initial promise as a rejuvenating factor before becoming the subject of considerable scientific controversy regarding its measurement and interpretation, and more recently the platelet-derived exerkine GPLD1. Aged plasma conversely contains elevated levels of factors including CCL11 (eotaxin), TGF-beta1, and beta2-microglobulin that have been shown to impair neurogenesis, muscle regeneration, and cognitive function when infused into young animals. The identification and therapeutic targeting of pro-aging circulating factors represents one of the most actively pursued areas in geroscience as of 2026.
Extracellular vesicles (EVs), including exosomes (30 to 150 nm) and microvesicles (100 to 1000 nm), have emerged as major vehicles of intercellular communication with documented roles in aging. EVs carry diverse molecular cargo including proteins, lipids, and non-coding RNAs, particularly microRNAs, that can alter gene expression in recipient cells. The EV cargo composition changes with age in ways that promote inflammation and impair regeneration, and EV-based delivery of specific molecular payloads is being explored as a therapeutic modality for age-related conditions.
The Aging Brain: Neurochemistry and Cognitive Decline
The brain occupies a uniquely vulnerable position in the context of aging biology. Neurons are post-mitotic cells that must be maintained for the entire lifespan of the organism, accumulating molecular damage without the option of diluting it through cell division. At the same time, the brain consumes approximately 20 percent of the body’s resting oxygen while accounting for only 2 percent of its mass, generating proportionally large amounts of mitochondrial ROS. The intersection of these factors makes the brain a site of particularly pronounced age-related molecular change.
Neuroinflammation mediated by activated microglia, the brain’s resident macrophages, is a central feature of brain aging. Microglia adopt an inflammatory state in response to accumulating DAMPs from dying neurons and degenerating myelin, amyloid-beta deposits, and signals from the aged systemic circulation including elevated IL-6 and TNF-alpha. Chronically activated microglia produce proinflammatory cytokines and reactive oxygen and nitrogen species that are directly neurotoxic and that impair synaptic function by interfering with glutamate clearance by astrocytes and by complement-mediated synapse elimination. Recent large-scale single-cell transcriptomic atlases of the human and mouse aging brain have identified distinct microglial substates with specific molecular signatures, including disease-associated microglia (DAM) characterised by upregulation of APOE, TREM2, and lysosomal genes, that are enriched in aging and neurodegenerative conditions.
Synaptic function declines with normal aging independently of overt neurodegeneration. Long-term potentiation (LTP), the synaptic mechanism underlying learning and memory, is impaired in aged hippocampal tissue in part because of altered NMDA receptor composition (a developmental shift from NR2B-containing to NR2A-containing receptors reduces calcium influx and downstream signalling), elevated levels of inhibitory Rho GTPase signalling that restrict dendritic spine dynamics, and reduced BDNF expression. The resulting cognitive changes in normal aging, affecting processing speed, working memory capacity, and episodic memory retrieval while largely sparing semantic memory and procedural knowledge, can be distinguished neurobiologically from the more severe and progressive deficits of Alzheimer’s and related dementias, though the molecular risk factors substantially overlap.
Toward Intervention: The Emerging Chemistry of Anti-Aging Therapeutics
The translation of mechanistic insights into the chemistry of aging into interventions that extend human healthspan is no longer a speculative enterprise. Multiple classes of compounds with well-defined molecular targets in aging pathways are at various stages of preclinical and clinical development, and the conceptual framework of geroscience, which proposes that targeting fundamental aging mechanisms will simultaneously reduce the risk of multiple age-related diseases, has begun to receive empirical support.
Senolytics, compounds that selectively induce apoptosis in senescent cells by targeting their characteristic dependencies on anti-apoptotic BCL-2 family members, represent the most clinically advanced class. Navitoclax (ABT-263) inhibits BCL-2, BCL-XL, and BCL-W and has shown efficacy in clearing senescent cells and improving physical function in aged mice. The combination of dasatinib (a tyrosine kinase inhibitor that disrupts senescent cell survival signalling) and quercetin (a flavonoid that inhibits PI3K and BCL-2 family members via polypharmacology) has shown reductions in circulating senescent cell burden and SASP markers in small human trials in pulmonary fibrosis and diabetic kidney disease. Senolytics targeting the p21 pathway or the integrin-FAK survival axis are in earlier development.
NAD+ repletion with NR or NMN, mTOR inhibition with rapamycin or next-generation rapalogs, AMPK activation with metformin, and sirtuin activation with NAD+ precursors or SIRT1-activating compounds (STACs) such as resveratrol and its more potent successors constitute additional mechanistic strategies under clinical investigation. The challenge across all these approaches is identifying biomarkers that sensitively reflect target engagement and biological aging rate in humans, enabling trials of manageable duration. Epigenetic clocks, measures of p16-positive cell burden in peripheral blood, circulating SASP factor panels, and composite biological age scores are all under evaluation as trial endpoints in the growing geroscience clinical pipeline.
Where the Chemistry of Aging Leads
The biology of aging has been transformed over the past three decades from a descriptive science into a mechanistic one. What once appeared as the inevitable, programmed decline of living systems is now understood as the compounded consequence of specific, identifiable, and in many cases experimentally reversible chemical and molecular events: the erosion of protective telomere caps, the accumulation of somatic mutations, the drift of epigenetic landscapes, the failure of proteostasis networks, the dysfunction of mitochondria, the chronic signalling of senescent cells, and the inflammatory remodelling of the tissue microenvironment. Each of these processes has a defined biochemistry, and each connects to the others through a web of feedback and feedforward interactions that amplifies the cumulative damage of time.
The scientific trajectory now points clearly toward intervention. The identification of conserved longevity pathways, the demonstration that aging phenotypes can be reversed in animal models by targeting specific molecular processes, and the launch of clinical trials explicitly designed to delay aging as a medical endpoint together represent a qualitative shift in what geroscience aspires to achieve. The chemistry behind growing old is no longer merely an object of intellectual curiosity: it is the substrate for one of the most consequential medical endeavours of the coming decades. Whether targeting any single hallmark, or more likely combinations of hallmarks acting synergistically, will produce meaningful extension of human healthspan will be answered by the clinical science of the 2020s and 2030s. What the molecular biology of aging has already established, beyond reasonable doubt, is that the question is worth asking with full experimental rigour.
References
- Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. “The Hallmarks of Aging.” Cell. 2013.
- Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. “Hallmarks of Aging: An Expanding Universe.” Cell. 2023.
- Blackburn, E. H., Greider, C. W., and Szostak, J. W. “Telomeres and Telomerase: The Path from Maize, Tetrahymena and Yeast to Human Cancer and Aging.” Nature Medicine. 2006.
- Hayflick, L., and Moorhead, P. S. “The Serial Cultivation of Human Diploid Cell Strains.” Experimental Cell Research. 1961.
- Harman, D. “Aging: A Theory Based on Free Radical and Radiation Chemistry.” Journal of Gerontology. 1956.
- Horvath, S. “DNA Methylation Age of Human Tissues and Cell Types.” Genome Biology. 2013.
- Lu, A. T., Quach, A., Wilson, J. G., et al. “DNA Methylation GrimAge Strongly Predicts Lifespan and Healthspan.” Aging. 2019.
- Campisi, J. “Aging, Cellular Senescence, and Cancer.” Annual Review of Physiology. 2013.
- Coppe, J. P., Desprez, P. Y., Krtolica, A., and Campisi, J. “The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression.” Annual Review of Pathology. 2010.
- Franceschi, C., Bonafe, M., Valensin, S., et al. “Inflamm-Aging: An Evolutionary Perspective on Immunosenescence.” Annals of the New York Academy of Sciences. 2000.
- Fontana, L., Partridge, L., and Longo, V. D. “Extending Healthy Life Span: From Yeast to Humans.” Science. 2010.
- Harrison, D. E., Strong, R., Sharp, Z. D., et al. “Rapamycin Fed Late in Life Extends Lifespan in Genetically Heterogeneous Mice.” Nature. 2009.
- Kenyon, C. J. “The Genetics of Ageing.” Nature. 2010.
- Ziegler, D. V., Wiley, C. D., and Velarde, M. C. “Mitochondrial Effectors of Cellular Senescence: Beyond the Free Radical Theory of Aging.” Aging Cell. 2015.
- Imai, S., and Guarente, L. “NAD+ and Sirtuins in Aging and Disease.” Trends in Cell Biology. 2014.
- Tchkonia, T., Zhu, Y., van Deursen, J., Campisi, J., and Kirkland, J. L. “Cellular Senescence and the Senescent Secretory Phenotype: Therapeutic Opportunities.” Journal of Clinical Investigation. 2013.
- Barzilai, N., Crandall, J. P., Kritchevsky, S. B., and Espeland, M. A. “Metformin as a Tool to Target Aging.” Cell Metabolism. 2016.
- Wyss-Coray, T. “Ageing, Neurodegeneration and Brain Rejuvenation.” Nature. 2016.
- Jaiswal, S., Fontanillas, P., Flannick, J., et al. “Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes.” New England Journal of Medicine. 2014.
- Kuilman, T., Michaloglou, C., Mooi, W. J., and Peeper, D. S. “The Essence of Senescence.” Genes and Development. 2010.
- Ulrich, P., and Cerami, A. “Protein Glycation, Diabetes, and Aging.” Recent Progress in Hormone Research. 2001.
- Schumacher, B., Pothof, J., Vijg, J., and Hoeijmakers, J. H. J. “The Central Role of DNA Damage in the Ageing Process.” Nature. 2021.
- Goodell, M. A., and Rando, T. A. “Clonal Hematopoiesis and the Aging Stem Cell.” Science. 2015.
- Bhanu, N. V., and Garcia, B. A. “Histone Modifications and the Aging Epigenome.” Epigenetics and Chromatin. 2022.
- Longo, V. D., and Mattson, M. P. “Fasting: Molecular Mechanisms and Clinical Applications.” Cell Metabolism. 2014.