How Antibiotic Resistance is Evolving
Antibiotic resistance stands as one of the most formidable and accelerating challenges in modern medicine and public health. What began as a predictable biological consequence of introducing antimicrobial agents into clinical practice has, over the course of several decades, matured into a complex, globally distributed crisis that threatens to reverse a century of infectious disease progress. The molecular mechanisms underlying resistance are ancient in evolutionary terms, but the unprecedented selective pressure exerted by mass antibiotic use has compressed what would naturally be a glacial adaptive process into a timescale measurable in years, and in some documented cases, in days. Understanding how resistance evolves requires engaging simultaneously with molecular biology, microbial ecology, epidemiology, and evolutionary genetics, because the phenomenon operates at every one of these levels at once.
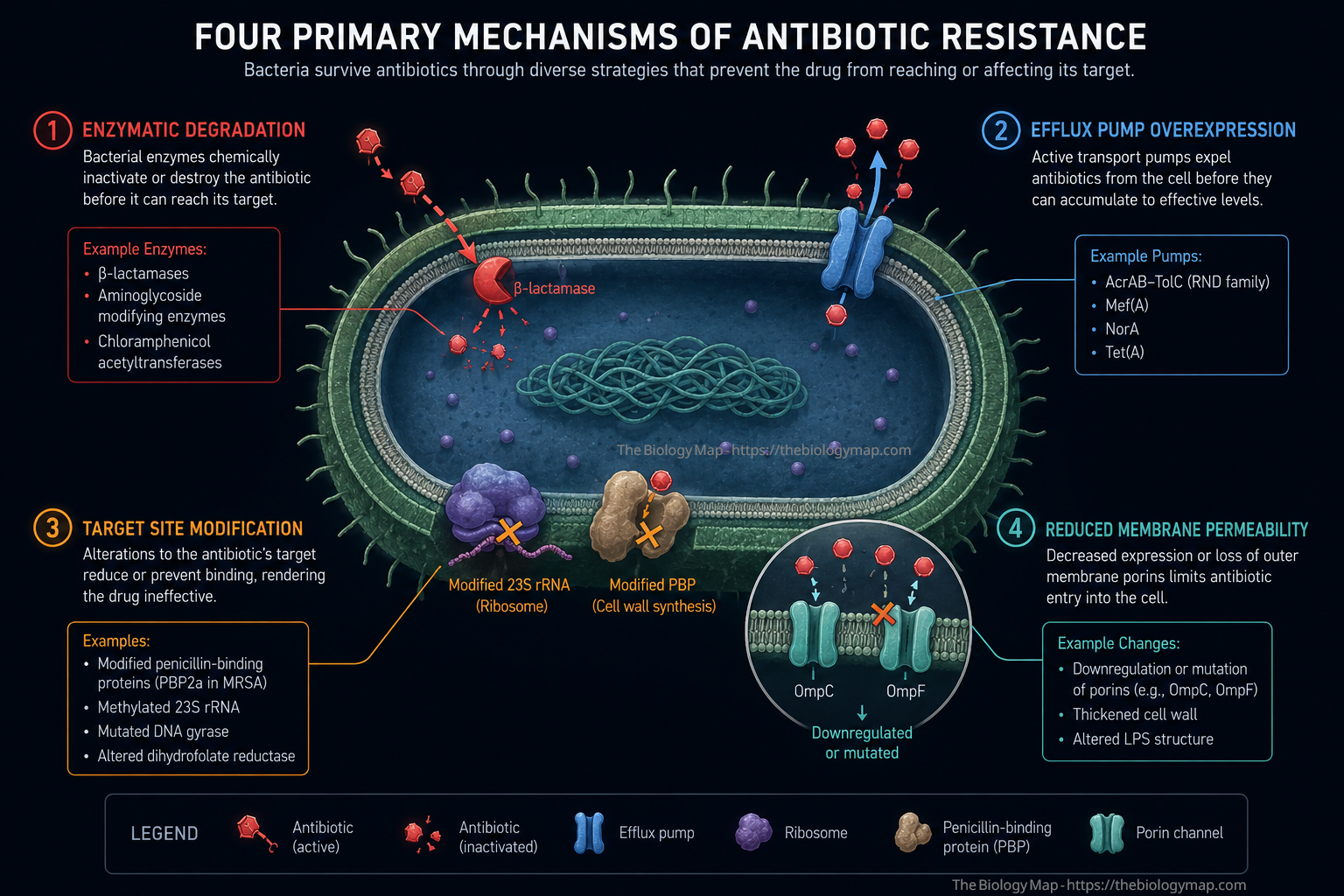
Figure 1: The four primary biochemical mechanisms of antibiotic resistance in bacterial cells. Enzymatic degradation (red) cleaves the antibiotic before it reaches its target; efflux pump overexpression (blue) actively expels the drug; target site modification (amber) prevents antibiotic binding to key cellular machinery such as ribosomes or penicillin-binding proteins; and reduced membrane permeability (teal) limits drug entry by downregulating porins such as OmpC and OmpF.
The Evolutionary Foundations of Resistance
Resistance to antimicrobial compounds is not a modern invention. Bacteria have coexisted with antibiotic-producing organisms, primarily Streptomyces species and other soil actinomycetes, for hundreds of millions of years. Environmental sampling studies have recovered functional resistance genes from permafrost cores and cave sediments entirely isolated from human activity, confirming that the biochemical toolkit for defeating antibiotics predates clinical medicine by an immense margin. What the antibiotic era has done is not create resistance so much as dramatically alter the selective landscape in which pre-existing resistance determinants propagate and diversify.
The foundational logic is Darwinian. A population of bacteria exposed to a bactericidal or bacteriostatic agent is subjected to intense directional selection. Sensitive cells are eliminated or growth-inhibited, while any cell carrying a heritable trait that confers even marginal survival advantage reproduces disproportionately. Over successive generations, the resistant phenotype comes to dominate the population. This process operates on spontaneous mutation rates that are low per gene per replication, approximately 10 to the power of negative 9 to 10 to the power of negative 10 per base pair per generation in Escherichia coli, but becomes highly consequential when population sizes reach the scale of 10 to the power of 8 to 10 to the power of 10 cells per millilitre in an infection site. Under those densities, every possible single-nucleotide variant at every position in the genome is being generated continuously, and positive selection acts with extraordinary efficiency to fix advantageous mutations.
What elevates bacterial resistance evolution far beyond a simple Darwinian model is the role of horizontal gene transfer (HGT), which allows resistance determinants to spread not only vertically from parent to daughter cell, but laterally between phylogenetically distant organisms in a single generation. The three principal mechanisms of HGT are transformation (uptake of naked DNA from the environment), transduction (bacteriophage-mediated gene transfer), and conjugation (direct cell-to-cell transfer via plasmids through a pilus structure). Of these, conjugation is epidemiologically the most consequential because it can transfer large, multi-resistance plasmids across genus and even phylum boundaries under conditions encountered in both clinical environments and the gastrointestinal tract.
Molecular Mechanisms: How Bacteria Defeat Antibiotics
The biochemical strategies that bacteria deploy against antibiotics are not randomly distributed. They fall into four well-defined mechanistic categories, each exploiting a different vulnerability in the drug-target relationship: enzymatic inactivation of the antibiotic, active efflux of the drug from the cell, modification of the antibiotic’s molecular target, and reduction of membrane permeability to limit drug entry in the first place.
Enzymatic Inactivation
Beta-lactamase enzymes represent the most clinically prevalent and historically significant class of resistance determinants. These enzymes hydrolyse the beta-lactam ring that is essential to the antibacterial activity of penicillins, cephalosporins, monobactams, and carbapenems. Beta-lactamases are classified by the Ambler scheme into classes A through D based on their catalytic mechanism: classes A, C, and D use a serine-based mechanism, while class B metallo-beta-lactamases employ zinc ions in the active site. The distinction matters clinically because current beta-lactamase inhibitors such as clavulanate, tazobactam, and avibactam are effective against serine-beta-lactamases but not against metallo-beta-lactamases, for which no inhibitor has yet achieved broad clinical deployment.
Extended-spectrum beta-lactamases (ESBLs), which hydrolyse third-generation cephalosporins, and carbapenemases, including the Klebsiella pneumoniae carbapenemase (KPC), New Delhi metallo-beta-lactamase (NDM), and OXA-48 families, have emerged as critical threats. The NDM-1 enzyme, first characterised in 2009 from a patient in Sweden with a history of hospitalisation in New Delhi, is now distributed across more than 70 countries and can be carried on highly mobile plasmids that co-transfer resistance to aminoglycosides, fluoroquinolones, and sulphonamides simultaneously. Beyond beta-lactamases, aminoglycoside-modifying enzymes, including acetyltransferases, phosphotransferases, and nucleotidyltransferases, chemically alter aminoglycoside antibiotics in ways that prevent them from binding with high affinity to the 30S ribosomal subunit, their primary target.
Efflux Pump Overexpression
Efflux pumps are membrane-embedded protein complexes that actively transport compounds, including antibiotics, from the cytoplasm or periplasm to the extracellular environment, using either proton motive force or ATP hydrolysis as the energy source. Five major superfamilies of efflux pumps have been characterised in bacteria: the resistance-nodulation-division (RND) family, the major facilitator superfamily (MFS), the ATP-binding cassette (ABC) superfamily, the small multidrug resistance (SMR) family, and the multidrug and toxin extrusion (MATE) family. The RND family is of particular clinical relevance in Gram-negative pathogens because it spans all three layers of the Gram-negative cell envelope via a tripartite complex, allowing direct expulsion of drugs into the external medium rather than merely the periplasm.
The MexAB-OprM system in Pseudomonas aeruginosa and the AcrAB-TolC system in E. coli are paradigmatic examples of RND-type pumps. Overexpression of these systems, achieved through mutations in local or global regulatory genes such as mexR, nalC, or marR, confers reduced susceptibility to structurally unrelated drug classes simultaneously, a phenomenon termed multidrug resistance (MDR). This is significant because resistance acquired through pump overexpression does not require separate mutational events for each antibiotic; a single regulatory mutation can simultaneously reduce susceptibility to beta-lactams, fluoroquinolones, tetracyclines, and chloramphenicol.
Target Site Modification
Antibiotics achieve their bactericidal or bacteriostatic effects by binding with high specificity and affinity to conserved bacterial structures, including the 30S and 50S ribosomal subunits, DNA gyrase and topoisomerase IV, penicillin-binding proteins (PBPs), RNA polymerase, and the lipid II cell wall precursor. Mutations or enzymatic modifications that alter the structure of these targets at or near the antibiotic binding site can drastically reduce drug affinity while preserving sufficient biological function to sustain bacterial viability.
Methicillin-resistant Staphylococcus aureus (MRSA) carries the mecA gene, which encodes PBP2a, a variant penicillin-binding protein with substantially reduced affinity for virtually all beta-lactam antibiotics. Because PBPs are the enzymes responsible for the terminal transpeptidation step in peptidoglycan synthesis, a bacterium expressing PBP2a retains cell wall biosynthetic capacity even in the presence of beta-lactam concentrations that would inactivate all native PBPs. The mecA gene resides on a mobile genetic element called the staphylococcal cassette chromosome mec (SCCmec), of which at least 13 structural types have been described, some carrying additional resistance and virulence determinants.
Fluoroquinolone resistance provides another instructive example. These drugs inhibit the DNA strand-passage reactions catalysed by DNA gyrase (a heterotetrameric A2B2 complex) and topoisomerase IV by forming a stabilised ternary complex with the enzyme and a double-stranded DNA break, ultimately triggering cell death. Resistance most commonly arises through single-amino-acid substitutions in the quinolone resistance-determining regions (QRDRs) of gyrA, gyrB, parC, and parE, the genes encoding the enzyme subunits. Each additional mutation in these QRDRs further increases the minimum inhibitory concentration (MIC), and organisms carrying multiple QRDR mutations may require fluoroquinolone concentrations far exceeding achievable serum levels. Plasmid-mediated quinolone resistance (PMQR) genes, including qnr variants and aac(6')-Ib-cr, further disseminate reduced fluoroquinolone susceptibility through HGT.
Reduced Membrane Permeability
In Gram-negative bacteria, the outer membrane constitutes a substantial permeability barrier that most hydrophilic antibiotics must traverse via proteinaceous channels called porins. The major porins of E. coli, OmpC and OmpF, form beta-barrel structures that allow passive diffusion of hydrophilic solutes up to approximately 600 daltons. Loss-of-function mutations or transcriptional downregulation of these porins reduces the rate of antibiotic accumulation within the periplasm and cytoplasm, thereby increasing effective MICs. When porin loss is combined with efflux pump overexpression, as is frequently observed in clinical carbapenem-resistant isolates of Klebsiella pneumoniae and Acinetobacter baumannii, the combinatorial effect can confer high-level clinical resistance even to antibiotics that partially retain activity against each mechanism in isolation.
Horizontal Gene Transfer and the Resistome
The concept of the resistome, introduced by Gerard Wright and colleagues, describes the totality of resistance genes and their precursors present in both pathogenic and non-pathogenic bacteria across all environments. Environmental metagenomics has dramatically expanded the catalogued size of the resistome, revealing resistance gene families in soil microbiomes, marine sediments, and the gut microbiota of wildlife with no history of anthropogenic antibiotic exposure. These environmental reservoirs serve as genetic libraries from which resistance determinants can be recruited into pathogenic lineages through HGT under selective pressure.
Integrons are perhaps the most efficient molecular machinery for assembling and disseminating resistance gene cassettes. Class 1 integrons, which are strongly associated with clinical resistance, consist of a platform with an integrase gene (intI1), a recombination site (attI), and a downstream promoter that drives expression of inserted gene cassettes. Individual cassettes, each consisting of a resistance gene and an associated recombination site (attC), can be excised and integrated in a site-specific recombination reaction, allowing multiple resistance cassettes to accumulate on a single integron. Class 1 integrons are frequently embedded within transposons, which in turn reside on conjugative plasmids, creating a nested, hierarchically mobile architecture that enables rapid resistance gene capture and inter-species dissemination.
Conjugative plasmids deserve particular attention as resistance vectors. The IncF, IncI, IncN, and IncX plasmid incompatibility groups have each been implicated in major resistance gene outbreaks. The epidemiology of plasmid-mediated resistance frequently follows a two-step pattern: a resistance gene first emerges or is acquired on a plasmid within a specific bacterial clone, and that clone then disseminates globally, after which the plasmid or resistance gene transfers horizontally to additional host species. The global spread of ESBL-producing E. coli of sequence type ST131, a fluoroquinolone-resistant uropathogenic clone that now dominates extraintestinal E. coli infections in multiple continents, exemplifies this pattern.
The Role of Selective Pressure in Accelerating Evolution
Selective pressure from antibiotic use is the proximal driver of resistance evolution, and the relationship between antibiotic consumption and resistance prevalence is documented at individual, institutional, and national levels. Countries with high per-capita antibiotic consumption consistently show higher rates of resistance in key pathogens. The World Health Organization’s Global Antimicrobial Resistance and Use Surveillance System (GLASS) data, updated through 2024, confirms that resistance to third-generation cephalosporins in E. coli and Klebsiella pneumoniae exceeds 50 percent in many low- and middle-income countries, correlating with patterns of antibiotic accessibility and regulation.
Sub-inhibitory antibiotic concentrations, those below the MIC of the pathogen, exert a less intuitively obvious but critically important selective pressure. At sub-MIC concentrations, resistant mutants are enriched relative to sensitive cells even in the absence of overt bactericidal effect. The concentration range between the MIC of the wild-type population and the MIC of the least-resistant mutant has been termed the mutant selection window (MSW). Pharmacokinetic-pharmacodynamic (PK-PD) modelling demonstrates that drugs with long tissue half-lives or those that distribute to poorly perfused compartments can expose bacterial populations to sub-MIC concentrations for extended periods, particularly at the end of a treatment course, creating conditions that preferentially enrich resistant subpopulations.
Agricultural antibiotic use constitutes a separate and quantitatively substantial selective pressure. Globally, the majority of antibiotic production by mass is consumed in food animal production, primarily for growth promotion and prophylaxis in intensive farming settings. The resistance genes selected in agricultural microbiomes are transferred to human-associated bacteria through multiple routes, including direct contact with animals or their environments, consumption of contaminated food products, and environmental dissemination through animal waste applied to soil or discharged to waterways. The colistin resistance gene mcr-1, located on a conjugative plasmid and conferring resistance to one of the last-resort polymyxin antibiotics, was first identified in food animals in China in 2015 and within months was detected in human clinical isolates across five continents, providing a near-real-time demonstration of the agricultural-to-human resistance transfer pathway.
Emerging and Evolving Resistance Threats in 2026
The landscape of clinically critical resistance continues to evolve along several trajectories that have accelerated since 2022. Extensively drug-resistant (XDR) and pandrug-resistant (PDR) isolates of the WHO priority pathogens, specifically carbapenem-resistant Acinetobacter baumannii (CRAB), carbapenem-resistant Pseudomonas aeruginosa (CRPA), and carbapenem-resistant Enterobacterales (CRE), now appear with sufficient frequency in tertiary care settings to constitute a genuine therapeutic emergency in affected regions.
The evolution of resistance within individual patients during treatment is an increasingly documented phenomenon. Whole-genome sequencing of serial isolates from patients with chronic P. aeruginosa infections in cystic fibrosis and bronchiectasis, or from patients receiving prolonged carbapenem therapy, reveals convergent adaptive trajectories: regulatory mutations in efflux pump systems, loss-of-function mutations in porin genes, and step-wise accumulation of target site mutations all occur on timescales of weeks to months within a single host. This within-host evolution is driven by the spatially structured, resource-heterogeneous environment of chronic infection, which creates diverse ecological niches and multiple simultaneous selective pressures.
Plasmid-mediated resistance to fosfomycin, a drug that had long been considered relatively resistant to plasmid-based resistance transfer, has now been described in clinical Enterobacterales isolates carrying the fosA gene on conjugative plasmids. The emergence of plasmid-mediated resistance to drugs previously considered impervious to such transfer reflects the extraordinary genetic plasticity of mobile resistance elements and suggests that the boundaries of “last-resort” antibiotic efficacy are narrower than historically assumed.
Resistance evolution in Mycobacterium tuberculosis follows a distinct trajectory because M. tuberculosis does not engage in HGT under natural infection conditions, relying instead entirely on chromosomal mutation for resistance acquisition. The global burden of multidrug-resistant tuberculosis (MDR-TB), defined as resistance to at least rifampicin and isoniazid, was estimated by the WHO at approximately 400,000 new cases annually as of 2024. Pre-extensively drug-resistant TB (pre-XDR-TB), now defined as MDR/RR-TB with additional resistance to any fluoroquinolone, represents the most rapidly growing resistance category, driven by the widespread use of fluoroquinolones both in TB treatment and in empirical management of community-acquired respiratory infections.
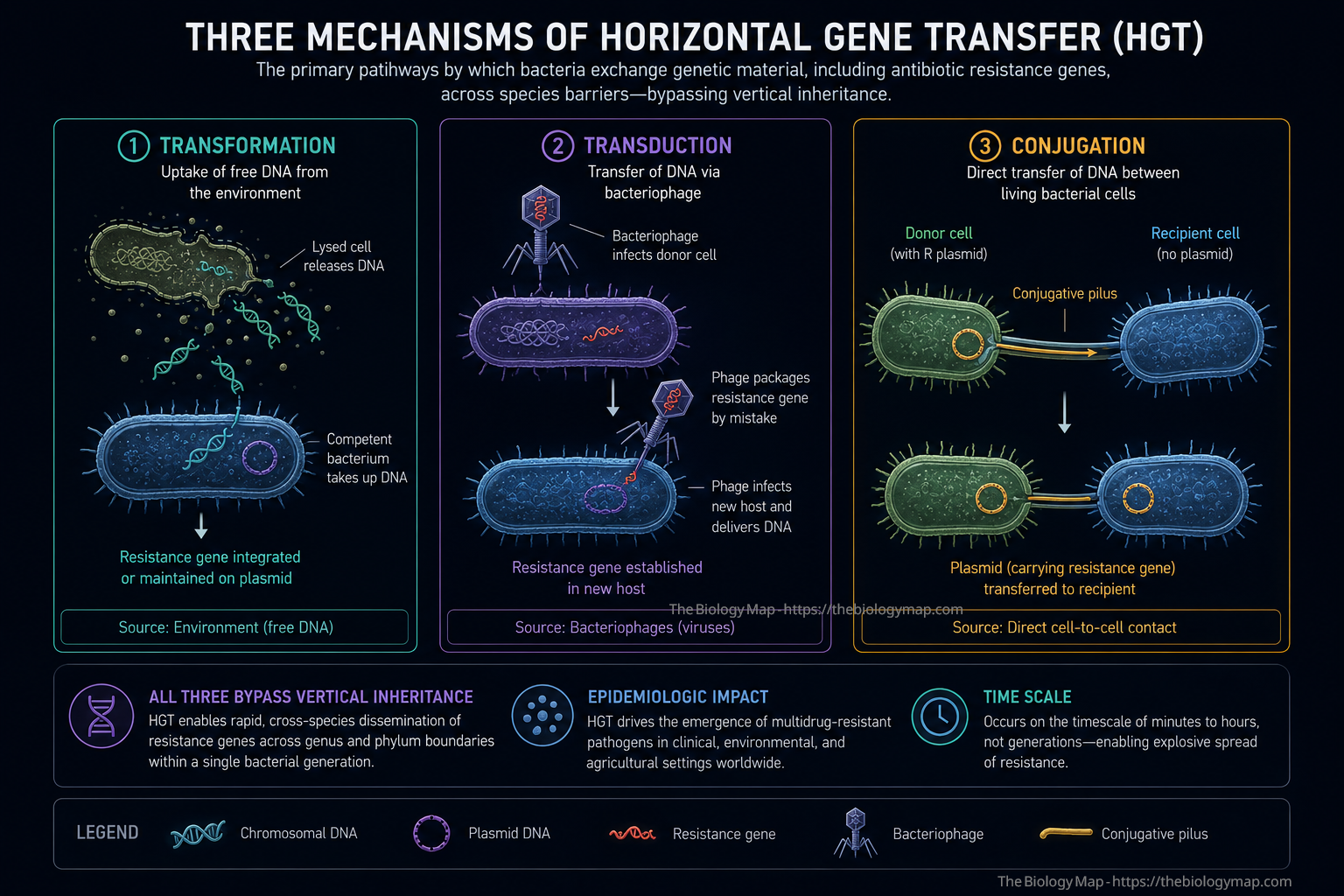
Figure 2: The three mechanisms of horizontal gene transfer (HGT) responsible for inter-species resistance gene dissemination. Transformation involves uptake of free environmental DNA from lysed cells; transduction uses bacteriophage as vehicles for resistance gene delivery; conjugation, the most epidemiologically significant mechanism, transfers resistance plasmids directly between live cells through a pilus structure. All three bypass vertical inheritance, enabling resistance to spread across genus and phylum boundaries within a single bacterial generation.
Within-Host Evolution and Adaptive Strategies
The within-host evolutionary dynamics of bacterial pathogens during infection and treatment are qualitatively distinct from population-level dynamics and have only become tractable to study with the widespread adoption of clinical whole-genome sequencing. Within the spatially structured microenvironment of an infected tissue or device-associated biofilm, bacterial subpopulations experience heterogeneous antibiotic concentrations, differential immune effector exposure, and localized nutrient gradients. These conditions create a complex, multi-niche fitness landscape on which adaptive evolution proceeds simultaneously along multiple axes.
Biofilm-associated infections are particularly instructive in this regard. Bacteria embedded in a self-produced extracellular matrix of polysaccharides, proteins, and extracellular DNA can tolerate antibiotic concentrations 10 to 1,000 times higher than their planktonic MIC, largely due to reduced drug penetration, altered metabolic states including slowed growth in the biofilm interior, and the presence of phenotypically antibiotic-tolerant “persister” subpopulations. Persisters are not genetically resistant; they are physiologically dormant cells that survive antibiotic exposure without heritable resistance mutations and resume replication when the drug is removed. However, persisters provide a reservoir of surviving cells within which de novo resistance mutations can accumulate during treatment, effectively converting phenotypic tolerance into durable genetic resistance through a two-step process.
Adaptive laboratory evolution studies and clinical genomic analyses converge on the observation that mutator phenotypes accelerate resistance evolution under antibiotic pressure. Bacteria with defective mismatch repair (MMR) systems, typically arising from mutations in mutS, mutL, or mutH, display genome-wide mutation rates 10 to 1,000 times higher than wild-type. Mutator lineages have been repeatedly recovered from chronic P. aeruginosa infections in cystic fibrosis patients and from patients with prolonged Gram-negative bacteraemia, suggesting that the clinical infection environment actively selects for elevated mutagenesis as a bet-hedging strategy that accelerates the generation of antibiotic-resistant variants.
The Interplay Between Resistance and Virulence
A historically prevalent assumption in clinical microbiology held that resistance mutations impose a fitness cost on bacteria, reducing growth rate or virulence relative to sensitive wild-type strains and thereby limiting the long-term epidemiological success of resistant lineages. While this is true for some resistance mutations in some genetic backgrounds, the assumption has proven far too simplistic as a general principle. Many resistant clinical lineages are not only epidemiologically successful but exhibit virulence characteristics at least equivalent to their sensitive counterparts.
Compensatory evolution, in which secondary mutations arise that restore fitness without reversing resistance, is now recognised as a central mechanism allowing resistant pathogens to stabilise their adaptive gains. In M. tuberculosis, rifampicin resistance most commonly arises from mutations in the rpoB gene encoding the RNA polymerase beta subunit. Many of these rpoB mutations impose measurable in vitro fitness costs, but clinical and experimental data demonstrate that compensatory mutations in other rpo genes, as well as in genes encoding RNA polymerase accessory factors, rapidly restore replication rate and transcriptional efficiency. Strains carrying both a resistance mutation and one or more compensatory mutations may be clinically indistinguishable in terms of virulence from fully drug-sensitive strains.
Furthermore, in some instances there appears to be direct mechanistic linkage between resistance and virulence. Efflux pump overexpression, for example, does not merely expel antibiotics; it also reduces intracellular concentrations of bile salts, fatty acids, and host-derived antimicrobial peptides, thereby increasing tolerance of conditions encountered during infection. Regulatory mutations that upregulate efflux pumps can simultaneously increase resistance and enhance survival within macrophages, suggesting that the clinical environment selects for a resistance-virulence co-adaptation rather than forcing a trade-off between the two.
Global Surveillance, Genomic Epidemiology, and the Challenge of Tracking Evolution
The global scale at which resistance evolves and disseminates demands surveillance infrastructure capable of detecting and characterising resistant strains with both speed and phylogenetic resolution. Traditional surveillance based on aggregate MIC data and susceptibility testing provides essential clinical information but cannot resolve questions about the origin, relatedness, and transmission pathways of resistant lineages. Whole-genome sequencing (WGS) has progressively addressed this gap and, as of 2025, is increasingly adopted for routine clinical microbiology in high-income countries as both a diagnostic and an epidemiological tool.
Genomic epidemiology, which applies phylogenetic and population genetic analysis to large collections of WGS data, has fundamentally redrawn the map of resistance evolution. The global phylogeny of MRSA, for example, reveals that the major healthcare-associated lineages (including USA100/CC8 and CC22-MRSA-IV) are geographically structured clonal expansions with distinct geographic adaptation and resistance gene carriage profiles, while community-associated MRSA lineages such as USA300 have demonstrated extraordinary adaptive radiation, acquiring resistance to tetracyclines, macrolides, and fluoroquinolones while retaining the virulence-associated Panton-Valentine leucocidin (PVL) locus. These insights are only accessible through WGS-based approaches.
The WHO Global Antimicrobial Resistance and Use Surveillance System (GLASS) coordinates national surveillance programmes across more than 100 member countries, but significant gaps remain in low- and middle-income country representation, which is particularly problematic given that these regions carry the highest burden of resistant infections and experience the most limited treatment options. Strengthening surveillance infrastructure in these settings is recognised as both an epidemiological and an ethical priority, since resistance that evolves and spreads in poorly monitored settings will inevitably affect global pathogen populations.
Metagenomic approaches offer a complementary surveillance strategy by enabling direct sequencing of resistance genes from environmental samples, hospital surfaces, wastewater, and gut microbiomes without the requirement for culture. Metagenomic surveillance of hospital wastewater has demonstrated the feasibility of tracking the emergence and dissemination of specific resistance gene variants in near-real time, providing an early-warning system for novel resistance events before they manifest as clinical treatment failures.
Strategies at the Intersection of Evolution and Intervention
Understanding the evolutionary dynamics of resistance opens conceptual space for intervention strategies that move beyond the traditional model of deploying new drugs against resistant pathogens and attempting to contain the resulting resistance through stewardship alone. Several evolutionarily informed strategies are under active investigation as of 2026.
Collateral sensitivity, also called evolutionary steering, exploits the observation that resistance to one antibiotic can increase susceptibility to another, creating a dependency between resistance genotypes and drug vulnerability. If drug A resistance carries collateral sensitivity to drug B, cycling or sequential use of these agents can theoretically drive the bacterial population into a fitness trap from which resistance cannot easily escape without reversing prior adaptations. Experimental evolution studies and mathematical models support the feasibility of collateral sensitivity exploitation, though translating this principle to complex, polymicrobial clinical infections with heterogeneous resistance profiles remains an active area of investigation.
Phage therapy, the therapeutic use of bacteriophages to kill bacterial pathogens, is experiencing a sustained revival driven by the availability of expanded phage libraries, synthetic biology tools for engineering phage host range and lytic efficiency, and an increasing number of compassionate use case reports demonstrating efficacy in XDR infections unresponsive to all available antibiotics. From an evolutionary perspective, a key advantage of phage therapy is that resistance to phage frequently carries fitness costs that restore antibiotic susceptibility, a phenomenon termed “phage-antibiotic synergy” when the two are co-administered. Clinical trials evaluating phage therapy in combination with antibiotics for P. aeruginosa and S. aureus infections are ongoing as of 2026.
Antivirulence strategies target bacterial pathogenesis mechanisms rather than growth per se, thereby exerting weaker selection for resistance while potentially neutralising infection. Inhibitors of quorum sensing systems, type III secretion systems, and toxin production are in various stages of preclinical and early clinical development. The evolutionary rationale is that drugs which do not kill bacteria or prevent growth do not create the intense directional selection that drives rapid resistance evolution. Whether antivirulence agents can achieve sufficient clinical efficacy without growth inhibition remains a central unresolved question.
Finally, the optimisation of existing antibiotic dosing regimens through rigorous PK-PD modelling represents an underutilised strategy for slowing resistance evolution. Dosing regimens designed to maintain drug concentrations above the mutant prevention concentration (MPC), the concentration above which resistant mutants cannot be selected from a population, reduce the probability of de novo resistance emergence during treatment. Extended-infusion and continuous-infusion dosing strategies for time-dependent antibiotics such as beta-lactams, which maximise the time that free drug concentrations exceed the MIC without increasing the total daily dose, are now endorsed in several international guidelines and are increasingly incorporated into clinical practice.
The Path Antibiotic Resistance Is Drawing for Medicine
The trajectory of antibiotic resistance evolution is not fixed, but the direction it is currently taking is unmistakably toward a narrowing of therapeutic options for an expanding category of infections. The convergence of molecular mechanisms that were once encountered separately, specifically the simultaneous presence of carbapenemases, ESBL genes, efflux pump overexpression, porin loss, and aminoglycoside-modifying enzymes in single clinical isolates, means that the concept of a “last-resort” antibiotic is losing meaningful definition for a subset of pathogens in specific geographic and clinical contexts.
At the same time, the scientific understanding of how resistance evolves has never been more sophisticated. The integration of evolutionary biology with microbiology, genomics, pharmacology, and ecology has produced a richer mechanistic framework than existed even a decade ago. This framework identifies intervention points that were previously invisible: the mutant selection window, the resistome, collateral sensitivity networks, within-host adaptive trajectories, and the agricultural-to-human resistance gene transfer pathway. Each of these represents not merely a conceptual insight but a potential target for evidence-based policy or therapeutic design.
Translating this understanding into durable clinical and public health impact requires coordinated action across several fronts simultaneously. Antimicrobial stewardship programmes must evolve from simple consumption reduction efforts to evolutionarily informed prescribing strategies that account for PK-PD optimisation, local resistance epidemiology, and collateral sensitivity. Regulatory frameworks governing agricultural antibiotic use must be strengthened and harmonised internationally, with particular urgency in the major livestock-producing nations where regulatory gaps remain largest. Investment in novel antibiotic discovery, alternative therapeutic modalities including phage therapy and antivirulence agents, and global surveillance infrastructure must proceed in parallel, since no single intervention is sufficient at the scale of the problem.
The evolutionary arms race between human medicine and bacterial adaptation is not a narrative that admits a final victory for either side. What it does admit is the possibility of an informed, sustained, and scientifically grounded effort to slow the pace of resistance evolution, preserve the efficacy of existing and future antibiotics, and maintain the foundation of effective infectious disease medicine that the antibiotic era built. The biology of resistance demands nothing less than the full mobilisation of that understanding.
References
- Wright, G.D. “The antibiotic resistome: the nexus of chemical and genetic diversity.” Nature Reviews Microbiology. 2007.
- Blair, J.M.A., Webber, M.A., Baylay, A.J., Ogbolu, D.O., and Piddock, L.J.V. “Molecular mechanisms of antibiotic resistance.” Nature Reviews Microbiology. 2015.
- Nikaido, H. “Multidrug resistance in bacteria.” Annual Review of Biochemistry. 2009.
- Davies, J., and Davies, D. “Origins and evolution of antibiotic resistance.” Microbiology and Molecular Biology Reviews. 2010.
- Liu, Y.Y., Wang, Y., Walsh, T.R., et al. “Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study.” The Lancet Infectious Diseases. 2016.
- Perez, F., and Van Duin, D. “Carbapenem-resistant Enterobacteriaceae: a menace to our most vulnerable patients.” Cleveland Clinic Journal of Medicine. 2013.
- Woodford, N., and Ellington, M.J. “The emergence of antibiotic resistance by mutation.” Clinical Microbiology and Infection. 2007.
- Drlica, K., and Zhao, X. “Mutant selection window hypothesis updated.” Antimicrobial Agents and Chemotherapy. 2007.
- Brussow, H. “Phage therapy: the Escherichia coli experience.” Microbiology. 2005.
- Hoiby, N., Bjarnsholt, T., Givskov, M., Molin, S., and Ciofu, O. “Antibiotic resistance of bacterial biofilms.” International Journal of Antimicrobial Agents. 2010.
- Levin, B.R., and Rozen, D.E. “Non-inherited antibiotic resistance.” Nature Reviews Microbiology. 2006.
- Antimicrobial Resistance Collaborators. “Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis.” The Lancet. 2022.
- World Health Organization. Global Antimicrobial Resistance and Use Surveillance System (GLASS) Report: 2024. WHO Press. 2024.
- Alekshun, M.N., and Levy, S.B. “Molecular mechanisms of antibacterial multidrug resistance.” Cell. 2007.
- Poole, K. “Efflux-mediated antimicrobial resistance.” Journal of Antimicrobial Chemotherapy. 2005.
- Mahon, B.M., Brehony, C., McGrath, E., et al. “Indistinguishable NDM-producing Escherichia coli isolated from humans and environment.” Journal of Antimicrobial Chemotherapy. 2017.
- Oliver, A., Canton, R., Campo, P., Baquero, F., and Blazquez, J. “High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection.” Science. 2000.
- Melnyk, A.H., Wong, A., and Kassen, R. “The fitness costs of antibiotic resistance mutations.” Evolutionary Applications. 2015.
- Imamovic, L., and Sommer, M.O.A. “Use of collateral sensitivity networks to design drug cycling protocols that avoid resistance development.” Science Translational Medicine. 2013.
- Cassini, A., Hogberg, L.D., Plachouras, D., et al. “Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and EEA in 2015: a population-level modelling analysis.” The Lancet Infectious Diseases. 2019.