Can AI Understand Life?
The question sounds almost philosophical, the kind posed in seminar rooms or late-night conversations rather than in the measured language of molecular biology. Yet in 2026, it sits squarely at the intersection of computational science and the life sciences, demanding a rigorous rather than a romantic answer. Artificial intelligence has, without question, transformed what biologists can predict, model, and design. It has solved problems that were considered intractable for decades, catalogued the structures of hundreds of millions of proteins, and begun to decode the regulatory grammar embedded in non-coding DNA. Whether any of that constitutes “understanding” is a question worth interrogating precisely, because the answer shapes how we deploy these tools, where we trust them, and what we still need to build.
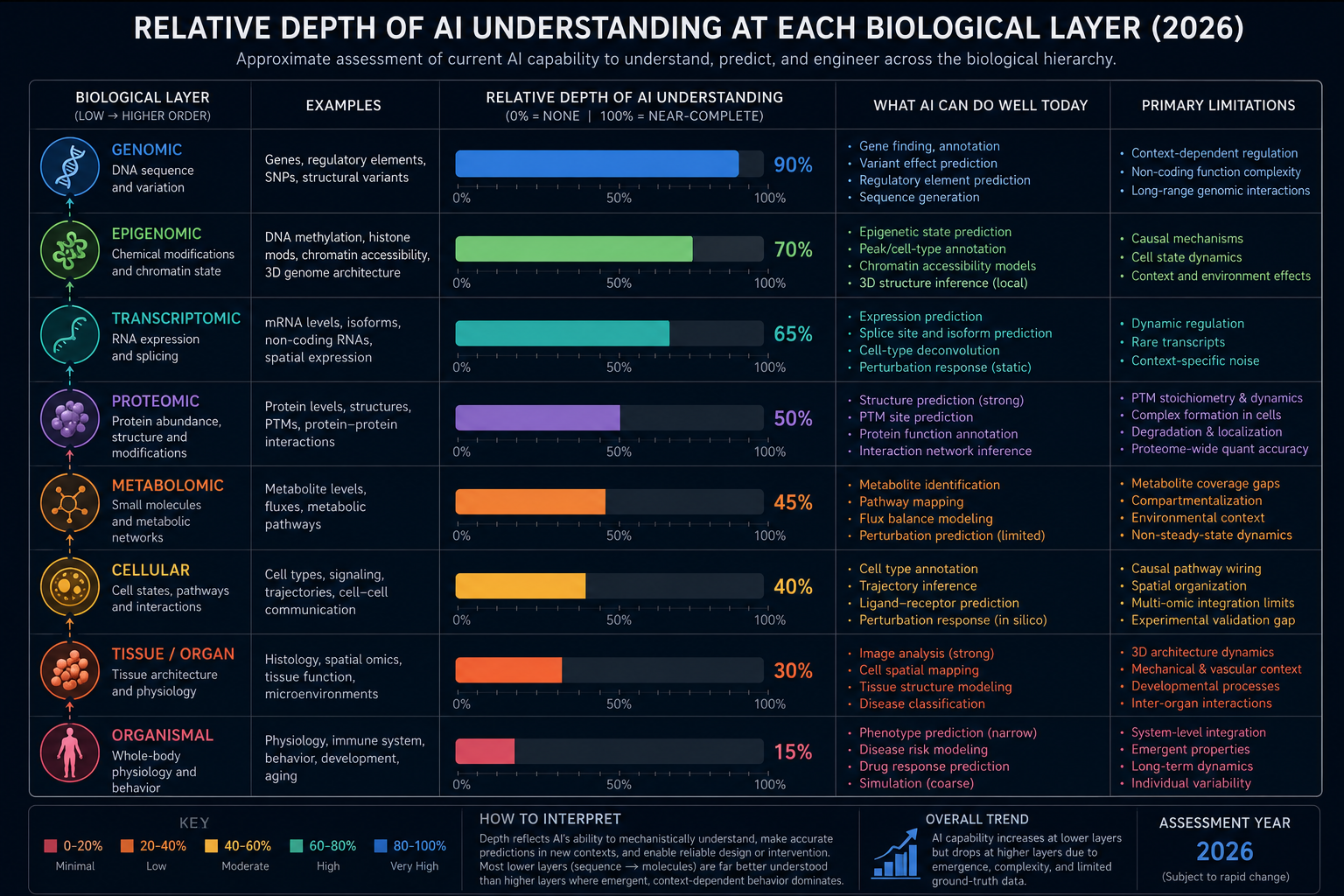
Approximate relative depth of AI understanding at each biological layer, 2026
The distinction matters enormously in practice. A system that predicts the correct output from a given input may or may not have captured anything meaningful about the mechanism producing that output. Biology is a field built on mechanisms: the sequence of molecular events that causally connects a genotype to a phenotype, a signal to a response, a mutation to a disease. When AI models reproduce the outputs of those mechanisms with remarkable accuracy, the temptation is to conclude that the mechanism itself has been learned. The evidence, examined carefully, suggests that the truth is considerably more complicated.
The Sequence Revolution: Where AI Excels Most Clearly
The most unambiguous successes of AI in biology have occurred at the level of sequence. Biological sequence, whether nucleotide or amino acid, encodes information in a linear, discrete, and digitally tractable format. This is, in a fundamental sense, the language that deep learning was built to read. The analogy between protein sequences and natural language, while imperfect in its mechanistic implications, is computationally productive: both involve discrete tokens arranged in strings where long-range dependencies matter, where context determines meaning, and where vast corpora of evolutionary or linguistic history provide training signal.
Protein language models represent the most mature expression of this approach. EvolutionaryScale’s ESM3, released in 2024, was trained across approximately 771 billion tokens of protein sequence, structure, and function data, building internal representations that capture evolutionary covariation across hundreds of millions of sequences. The model can generate novel proteins with specified structural and functional properties, reason across modalities simultaneously, and produce outputs that fold correctly when synthesised experimentally. This is not a trivial achievement. The evolutionary sequence space for a protein of typical length is astronomically large: Cyrus Levinthal’s 1969 calculation, now a classic illustration of the problem, estimated that exhaustive search of conformational space would require longer than the age of the known universe. ESM3 and its contemporaries navigate that space with a fluency that suggests genuine compression of evolutionary information.

From amino acid sequence to predicted structure The AlphaFold / ESM paradigm, with key constraint noted
DeepMind’s AlphaFold series represents the most celebrated instantiation of this capability. AlphaFold2, released in 2021, achieved near-experimental accuracy in predicting single-chain protein structures, effectively solving a fifty-year challenge in structural biology and earning the 2024 Nobel Prize in Chemistry. Its successor, AlphaFold3, extended predictions to protein-ligand, protein-nucleic acid, and protein-protein complexes using a diffusion transformer architecture that reasons over atomic coordinates rather than residue representations alone. In benchmark datasets, AlphaFold3 achieved 76.4 percent accuracy in protein-ligand docking, representing a 1.8-fold improvement over prior computational approaches and outperforming physics-based methods by approximately 50 percent. The evolutionary data informing these models is staggering in scale: multiple sequence alignments drawing on databases of over 650 million protein sequences distilled from billions of years of organismal evolution.
At the genomic level, AI models trained on regulatory DNA sequences have demonstrated the capacity to predict transcription factor binding sites, chromatin accessibility, and the functional consequences of non-coding variants with accuracy that substantially exceeds earlier computational methods. Tools including DeepBind, Enformer, and their successors have revealed that the cis-regulatory code, the set of short sequence motifs and their combinatorial arrangements that determine when and where genes are expressed, is learnable from sequence alone to a degree that surprised many researchers. These models have identified thousands of variants in non-coding regions that were previously difficult to prioritise, accelerating both mechanistic genomics research and the identification of candidate therapeutic targets.
Foundation Models for Biology: A Rapidly Expanding Ecosystem
The proliferation of biological AI models between 2015 and 2026 has been exponential in the most literal sense. Fewer than ten new biology-focused AI models were released annually in 2015; by 2024, that number had exceeded 350. The ecosystem now includes tools purpose-built for tasks spanning generative protein design, genomic perturbation modelling, single-cell transcriptomics, and histopathology image analysis. This diversity of application domains reflects a broader transition in how the field conceptualises computational biology: not as a collection of task-specific bioinformatics pipelines, but as an infrastructure of general-purpose foundation models adaptable to downstream biological questions.
Several models have achieved particular prominence by virtue of either their architectural novelty or their demonstrated utility. Boltz-2, announced in June 2025 by researchers at the MIT Jameel Clinic in collaboration with Recursion Pharmaceuticals, extends the diffusion architecture of AlphaFold3 by jointly modelling protein-ligand complex structures and binding affinities, incorporating a continuous affinity score analogous to a log IC50 measurement alongside standard coordinate prediction. This joint modelling of structure and binding energy is mechanistically significant, because the two properties are coupled in ways that sequential prediction pipelines cannot capture. Chai-1, from Chai Discovery, achieves multimodal biomolecular structure prediction across diverse molecule classes. The Evo model, trained on DNA sequences across prokaryotic genomes, enables prediction of the functional consequences of mutations at a genome-wide scale, including in non-coding regions not previously amenable to systematic computational analysis.
scGPT represents a parallel development in single-cell genomics, applying transformer architectures to single-cell RNA sequencing data to learn gene-gene relationships and cell-state representations in an unsupervised fashion. The model can be fine-tuned for tasks including cell-type annotation, gene regulatory network inference, and perturbation prediction, providing a flexible computational substrate for the analysis of cellular heterogeneity that is otherwise difficult to systematise. The commercial consequences of this ecosystem have been significant and accelerating. In early 2026, GSK committed $50 million upfront to NOETIK for access to oncology foundation models, while Eli Lilly paid a mid-eight-figure annual fee to Chai Discovery for biologics design capabilities. In April 2026, Anthropic acquired Coefficient Bio, a startup founded by former computational biologists from Evozyne, Genentech, and Prescient Design, for $400 million in equity, marking the entry of frontier AI laboratories into direct drug discovery investment.
The pace of model development raises an important caution that is easy to overlook amid commercial enthusiasm. The number of published models is not equivalent to the number of validated, mechanistically interpretable models. A substantial fraction of the 350-plus models catalogued by 2024 addressed the same small set of well-benchmarked tasks, such as structure prediction and binding classification, where rich experimental ground truth is available. The biological questions that are hardest to answer are precisely those for which experimental ground truth is sparse, temporally dynamic, or context-dependent in ways that resist simple formalisation.
The Structure-Function Gap: What Prediction Cannot Deliver
The most significant limitation of current biological AI models is one that becomes clear only when the excitement over benchmark performance is set aside. Predicting the three-dimensional structure of a protein from its sequence, as AlphaFold3 does with remarkable accuracy for many targets, provides a static snapshot of a molecular machine in one of the many conformational states it occupies during its biological lifetime. Living proteins are not static. They undergo conformational changes upon ligand binding, post-translational modification, interaction with partner proteins, and shifts in the cellular environment. The allosteric communication between distant sites on a protein, the coupling of structural dynamics to catalytic function, and the modulation of activity by cellular context are all mechanistic properties that a static structure prediction cannot directly address.
This limitation is not merely theoretical. Systematic benchmarking of AlphaFold3 in drug discovery applications has revealed that the model struggles significantly with protein-ligand complexes involving conformational changes greater than 5 angstroms root-mean-square deviation (RMSD). It displays a persistent bias toward active G-protein coupled receptor (GPCR) conformations, limiting its utility for the substantial portion of GPCR-targeting drug programmes that must account for inactive-state structures and state-dependent ligand binding. Most critically, AlphaFold3 shows minimal correlation between its predicted structural outputs and experimental binding affinities, which are the quantities that actually determine whether a candidate compound will be therapeutically useful. The model predicts where a ligand sits; it does not reliably predict how tightly it binds.
These limitations are not failures of the AlphaFold approach per se; they reflect a principled distinction between structure prediction and function prediction that has been recognised in structural biology for decades. The structure-function relationship in proteins is not a simple bijection. Many proteins adopt multiple functionally relevant conformations, and the relative populations of those conformations under physiological conditions, a quantity governed by the free energy landscape of the molecule in its cellular environment, are invisible to a model trained to output a single predicted structure. The introduction of Boltz-2’s joint structure-affinity modelling represents an important step toward bridging this gap, but it addresses binding affinity for small-molecule ligands rather than the full complexity of biological function in cellular context.
The challenge of biological dynamics extends beyond protein conformation. Gene regulatory networks are not static circuits but temporally dynamic systems whose behaviour depends on the history of inputs, the availability of transcription factors in a given cell type, the chromatin accessibility landscape at a particular developmental stage, and the autocrine and paracrine signals present in a tissue. Single-cell foundation models such as scGPT learn representations of gene co-expression relationships that are powerful for classification and interpolation tasks, but their capacity to predict the consequences of transcriptional perturbations in novel cellular contexts, particularly those not well represented in training data, remains limited and context-sensitive in ways that restrict generalisation.
Emergence, Context, and the Hard Problem of Biological Complexity
Understanding life at a mechanistic level requires engaging with emergence, the property by which complex behaviours arise from the interactions of simpler components in ways that cannot be straightforwardly predicted from the properties of those components in isolation. Emergence is not mystical; it is a well-defined concept in complex systems theory. But it is computationally demanding in a specific way: emergent properties are not reducible to local rules applied to individual components, and they are typically sensitive to the global organisation of the system in ways that resist description by any finite set of pairwise interactions.
The cell is an archetypal emergent system. Its capacity for homeostasis, for self-repair, for division, for differentiation, and for coordinated response to environmental perturbation arises from thousands of molecular species interacting across multiple spatial scales and temporal regimes simultaneously. A model that has learned the structures of all constituent proteins, the binding affinities of all pairwise interactions, and the regulatory logic of all individual gene circuits still faces an enormous inferential distance between that knowledge and a predictive account of cell-level behaviour. The epistemic challenge is not merely computational scale but the nature of the causal relationships involved: nonlinear feedback, multistable dynamics, and the role of molecular noise in shaping population-level outcomes.
Current AI models in biology are, for the most part, trained on correlational data. They learn that sequences with certain properties tend to produce structures with certain geometries, that expression patterns with certain characteristics tend to co-occur with certain cell types, that genomic variants with certain features tend to be associated with certain phenotypes. These correlational models are extraordinarily useful for many practical purposes, but they do not, in general, capture causal mechanisms in the biological sense. The distinction between correlation and causation is not merely philosophical in this context: a model that has learned correlations without causal structure will fail systematically when applied to interventional settings, precisely the settings most relevant to therapeutics, where the goal is to change a biological outcome by perturbing a specific molecular target.
This distinction has been made explicit by researchers calling for explainable AI in protein science. A perspective published in Nature Machine Intelligence in May 2026, authored by researchers at the Centre for Genomic Regulation in Barcelona, argued that protein language models have advanced the predictive power of computational biology while simultaneously reducing its transparency. Where physics-based models offered mechanistic accounts of folding energetics that could be interrogated and challenged, deep learning models offer predictions whose internal representations resist straightforward interpretation. The authors noted that understanding fundamental biological processes such as folding or catalysis has not advanced alongside the predictive breakthroughs, and that without better methods for explaining what models learn and how they make decisions, the field risks building powerful tools that it cannot fully trust or mechanistically validate.
Multi-Omics Integration and the Promise of Systems-Level Learning
One of the most promising frontiers in biological AI is the integration of multiple data modalities, genomics, transcriptomics, proteomics, metabolomics, epigenomics, and phenotypic imaging, into unified models capable of reasoning across molecular layers simultaneously. The rationale is mechanistically sound: biological function is not encoded at any single omics level but emerges from the coordinated activity of processes spanning all of them. A transcriptional response to a signalling event involves changes in chromatin accessibility, binding of transcription factors to newly accessible regulatory elements, altered RNA polymerase recruitment, post-transcriptional regulation of transcript stability and translation efficiency, and ultimately changes in the composition and activity of the proteome.
Models trained exclusively on sequence data or exclusively on structural data capture only one cross-section of this causal cascade. Multi-omics integration approaches aim to learn the cross-modal relationships that connect these levels, using the correlational structure present in large-scale datasets as a proxy for the mechanistic relationships that actually govern cellular behaviour. Projects such as insitro, which pairs large-scale human cell phenotypic data generation with machine learning across multiple omics modalities, have demonstrated that this approach can yield biologically meaningful insights: Bristol Myers Squibb’s nomination of two additional amyotrophic lateral sclerosis (ALS) targets from an insitro collaboration in 2026 provides one concrete clinical-stage validation of the approach.
The single-cell revolution has been particularly enabling for multi-omics modelling. Technologies including single-cell ATAC sequencing, CUT&Tag, and spatial transcriptomics now permit the simultaneous measurement of multiple molecular layers within individual cells and their tissue contexts, providing the ground truth data that multi-modal foundation models require. The scGPT framework and related tools can be conditioned on multiple input modalities, learning joint embeddings that capture the co-variation of chromatin state, gene expression, and cell identity within the same latent space. Whether these learned embeddings encode causal biological relationships or sophisticated correlational summaries is, again, a question that benchmarking on held-out prediction tasks cannot fully resolve.
The honest assessment of multi-omics AI is that it excels at discovering patterns in data that humans would not have detected by inspection, and that some of these patterns correspond to real biological mechanisms. It is less reliable as a tool for mechanistic discovery when applied to phenomena outside its training distribution, or when the relevant biology is governed by rare cell states, tissue-level organisation, or temporal dynamics not well represented in existing datasets. The field’s trajectory is toward larger datasets and more comprehensive multi-modal measurement, which will progressively improve coverage; but the question of whether increased data scale can substitute for mechanistic prior knowledge remains actively debated.
What It Means to “Understand” in Biology
The framing of the original question, “can AI understand life,” requires that we be precise about what understanding means in the context of a scientific discipline. Understanding in biology has historically been defined in mechanistic terms: an explanation is satisfying when it identifies the molecular actors, their interactions, the causal sequence of events, and the quantitative parameters that govern the process of interest. This is the standard articulated in the classic reductionist programme of twentieth-century molecular biology, and it remains the benchmark against which explanatory claims are evaluated.
By this standard, current AI models do not understand the biological processes they model, in the same sense that a sophisticated weather prediction model does not understand fluid dynamics. Both systems can produce accurate predictions, and both systems encode information about the systems they model, but neither contains an explicit, inspectable, causally transparent account of the mechanisms responsible for the outputs they generate. The internal representations of a transformer model trained on protein sequences encode something about the evolutionary and biophysical constraints on protein sequence space, but that encoding is distributed across billions of parameters in a form that is not directly interpretable in biochemical terms.
This is not a counsel of despair. It is a clarification of what kind of tool biological AI currently is: an extraordinarily powerful pattern recognition and prediction system that operates on biological data, whose outputs are most reliable when evaluated within the distribution of its training data, and whose mechanistic interpretability is a subject of active research rather than an established capability. The gap between prediction accuracy and mechanistic understanding is real, but it is also tractable. Methods for probing the internal representations of biological foundation models, including activation analysis, causal interventions on model components, and comparisons between model-learned features and experimentally established biochemical properties, are beginning to provide windows into what these models have and have not captured.
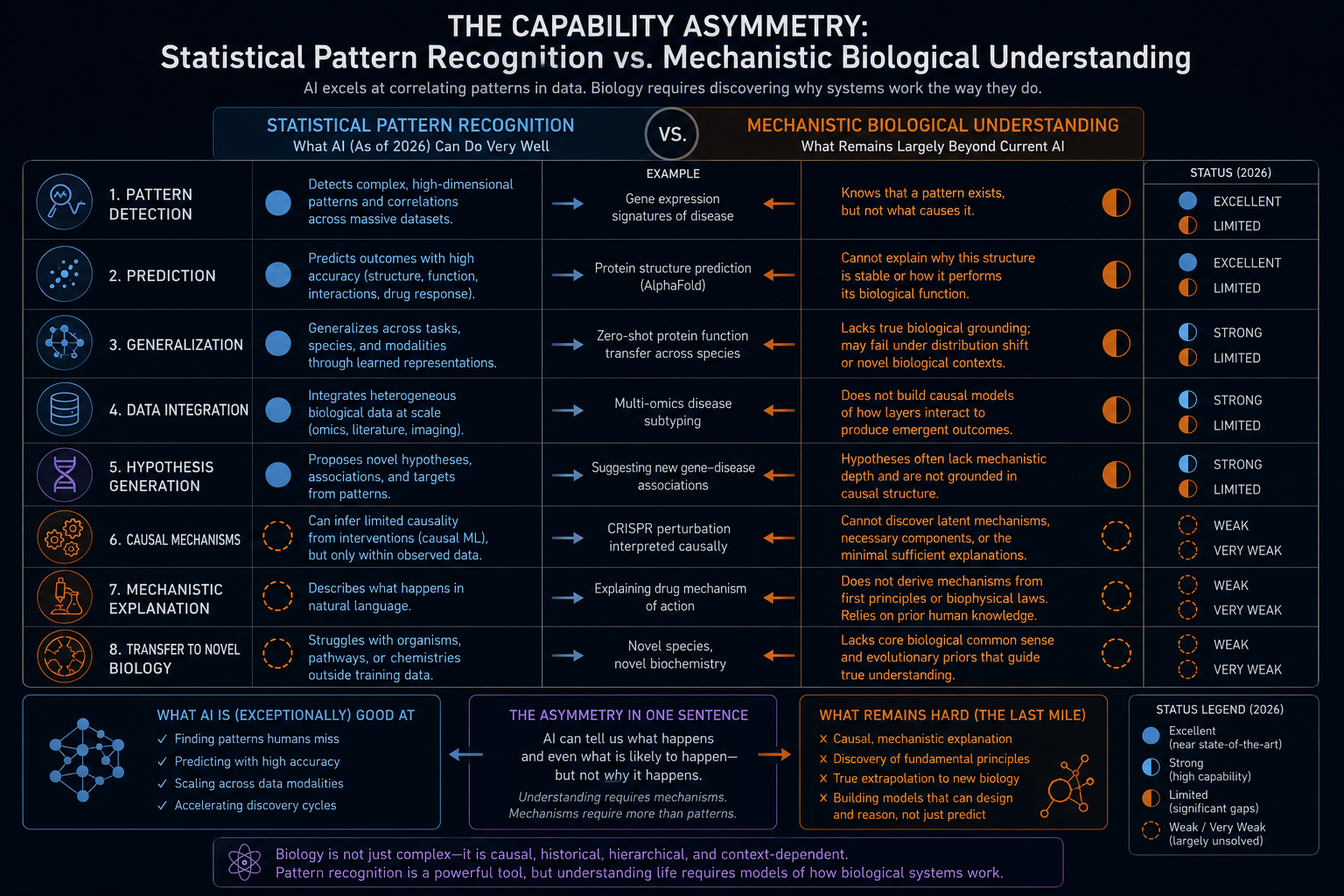
The capability asymmetry between statistical pattern recognition and mechanistic biological understanding
The broader question of whether any computational system, trained on biological data, could in principle acquire something analogous to the understanding that an experienced biologist develops through years of experimental practice is a philosophically non-trivial question. The biologist’s understanding is not purely propositional; it incorporates intuitions about biological plausibility, sensitivity to experimental artefacts, awareness of the contextual fragility of many molecular mechanisms, and a kind of embodied familiarity with biological systems developed through direct experimental engagement. Whether these dimensions of understanding are in principle transferable to a computational system is a question that current AI capabilities cannot resolve, and that the philosophy of science has not yet settled.
The Consciousness Question: A Separate and Harder Problem
The question of whether AI can understand life acquires an additional and more philosophically fraught dimension when it is applied not just to molecular and cellular biology but to the biology of mind. Consciousness, and the subjective experience of being a living organism, is the aspect of life that is most clearly inaccessible to current AI modelling, not merely as a practical limitation but as a matter of deep theoretical uncertainty.
The Cogitate Consortium’s adversarial collaboration results, published in Nature in April 2025, represented a significant moment in consciousness research: a pre-registered, multi-site collaboration designed to directly test the Integrated Information Theory and Global Neuronal Workspace Theory of consciousness against each other using shared data found that neither theory’s predictions were fully supported, destabilising the two most prominent neuroscientific frameworks that had aspired to a complete mechanistic account of conscious experience. The subsequent articulation of “biological computationalism” in December 2025, a position arguing that the physical, metabolic, and dynamic properties of biological tissue are constitutive rather than merely incidental to conscious experience, further complicated the landscape for those who had assumed that substrate-independence would allow consciousness to be realised in silicon as readily as in carbon.
Research published in Neuroscience and Biobehavioral Reviews in late 2025 by the Estonian Research Council argued specifically that consciousness cannot be reduced to its computational description, that the metabolic embedding of neural processing in living tissue contributes properties to conscious experience that a purely computational account cannot capture. Counterbalancing this, a philosopher at the University of Cambridge, Dr. Tom McClelland, argued in December 2025 that there is currently no evidence sufficient to establish either that consciousness requires biological substrate or that the right computational organisation is sufficient to produce it. The honest scientific position, he suggested, is that the field remains an intellectual revolution away from any viable test for consciousness in non-biological systems.
What is clear from this landscape is that the question of whether AI understands life in the deepest sense, the sense that involves understanding what it is to be a living, experiencing organism, is not currently answerable by any empirical method available to the field. The tools that enable AI to predict protein structures, identify regulatory variants, and design novel biomolecules with impressive accuracy operate at a level of abstraction far removed from the biology of subjective experience. Whether closing the gap between those levels is a matter of scale, architecture, data, or something more fundamental remains one of the most important and genuinely open questions in contemporary science.
Toward a Biologically Grounded AI
The limitations of current biological AI are not arguments against its development or deployment; they are arguments for developing it with appropriate epistemic humility and with sustained investment in the experimental biology that can validate, challenge, and ultimately ground its outputs. The most productive relationship between AI and biological science is not one in which AI replaces mechanistic investigation but one in which it accelerates hypothesis generation, narrows experimental search spaces, and reveals patterns in data that would otherwise remain invisible, with experimental biology providing the causal validation that statistical learning alone cannot supply.
Several developments point toward a more biologically grounded AI. The integration of physical constraints into model architectures, incorporating thermodynamic principles, known kinetic parameters, and mechanistic biochemical models as inductive biases rather than leaving all structure to be learned from data, is an active area of research that may improve both accuracy and interpretability outside training distributions. The development of foundation models trained jointly on perturbational data, measuring the consequences of systematic genetic or pharmacological interventions rather than correlational observational data alone, offers a path toward models that learn something closer to causal biological structure. The emerging field of AI-guided experimental design, in which AI models propose the most informative experiments to resolve mechanistic uncertainties, rather than simply predicting outcomes of known conditions, represents a potentially transformative integration of computational and experimental biology.
Bio-inspired AI itself offers a complementary direction. Research from Dehghani, Levin, and colleagues has argued that fundamental principles from biological computation, including context-dependent and hierarchical information processing, trial-and-error heuristics operating across multiple timescales, and multi-scale organisational structure, are largely absent from current deep learning architectures, and that incorporating them may be necessary for AI systems to achieve the kind of flexible, generalising intelligence that biological organisms display. This is not a claim about consciousness; it is a claim about the architectural features that allow biological neural systems to handle novelty, integrate information across spatial and temporal scales, and maintain adaptive behaviour in the face of perturbation. Whether replicating these features computationally is necessary for AI to truly understand life, or merely sufficient to make it more practically useful, is itself a question worth investigating rather than assuming.
Beyond the Benchmark: What Understanding Biologically Requires
The benchmark-driven culture of AI development creates systematic pressures that are worth examining critically in the biological context. A model that achieves state-of-the-art performance on the CASP protein structure prediction benchmark, the PDBbind docking benchmark, or the ENCODE regulatory element prediction benchmark has demonstrated something genuinely impressive and scientifically valuable. But benchmarks necessarily operationalise understanding as prediction accuracy on a defined task, and the tasks that can be cleanly benchmarked are not representative of the full range of biological understanding that the field requires.
The biological questions most consequential for human health, for ecological stability, and for our scientific understanding of life, including how mutations in regulatory DNA cause developmental disorders through altered gene dosage in specific cell lineages, how metabolic rewiring in cancer cells enables immune evasion, how ecosystems respond to combined stressors that have not previously co-occurred in evolutionary history, are precisely those for which clean benchmark datasets do not exist and may never exist in a form that current AI evaluation frameworks can accommodate. Progress on these questions requires not just better models but better integration between modelling and experimental investigation, sustained over time by research programmes that prioritise mechanistic understanding alongside predictive performance.
The trajectory of biological AI in 2026 is one of genuine and accelerating capability, accompanied by a growing recognition among the most careful practitioners that capability and understanding are not synonyms. The field has learned to predict with extraordinary power; the question of whether it has begun to understand remains, appropriately, a matter of active scientific investigation rather than settled conclusion.
Intelligence Without Experience: The Limits of Knowing Life from the Outside
There is a dimension of biological understanding that may be inaccessible to any system that does not itself inhabit a living body, subject to evolutionary pressures, metabolic constraints, and the vulnerability and finitude that characterise all biological existence. This is not a mystical claim but a scientific hypothesis worth taking seriously in light of what we know about the relationship between biological organisation and the properties of living systems.
A Royal Society paper published in May 2026, authored by Safron, Levin, and collaborators spanning MIT, Tufts University, and multiple computational neuroscience programmes, argued that the temporospatial alignment of dynamic relations between a system and its world, the way in which biological organisms entrain their internal dynamics to external environmental cycles across multiple timescales simultaneously, may be a constitutive feature of the kind of intelligence that can genuinely model and respond to the world rather than merely approximate patterns within it. Large language models receive feedback through post-training fine-tuning and reinforcement learning, the paper noted, but this is of a different kind from the moment-to-moment temporal coupling that allows biological organisms to self-organise in adaptive response to their environments. Whether this distinction implies a fundamental ceiling on AI biological understanding or merely a current architectural limitation is a question the paper posed without resolving.
What this line of argument suggests, at minimum, is that the question of whether AI can understand life is not exhausted by asking whether it can predict biological outputs accurately. It also requires asking whether the kind of understanding that biology as a science aspires to, causal, mechanistic, contextual, and ultimately grounded in the nature of living systems as self-organising, thermodynamically open, and historically constituted entities, is in principle achievable by systems that stand outside biological existence. That question is not answered by benchmark performance. It may not be answerable by the tools currently available to either AI research or the philosophy of biology. It is, however, one of the most important questions that the convergence of artificial intelligence and the life sciences has placed before us, and it deserves to be asked with the same rigour that we apply to the molecular mechanisms we are only beginning to learn to predict.
The Question Remains Open, and That Is the Point
The answer to “can AI understand life” is not yes or no, and it would be intellectually dishonest to offer either unqualified response. What AI can do, with increasing sophistication and at expanding biological scales, is recognise patterns in biological data, generate hypotheses, predict molecular properties, and accelerate the pace of experimental inquiry in ways that were impossible a decade ago. These are not trivial achievements, and their practical consequences, in drug discovery, genomic medicine, synthetic biology, and ecological modelling, are already visible and growing.
What AI cannot yet do, and what may require more fundamental advances in both AI architecture and our scientific understanding of biological complexity, is capture the causal, dynamic, contextual, and emergent nature of living systems in a form that constitutes mechanistic understanding in the full sense that biology demands. The gap between statistical pattern recognition and causal mechanistic knowledge is real. The gap between molecular prediction and cellular, organismal, and ecological comprehension is real. The gap between any computational system and the subjective experience of being a living organism is, for now, a gap whose character we cannot fully characterise.
The productive response to this situation is not to deflate the genuine achievements of biological AI, nor to overclaim understanding where prediction is what has actually been demonstrated. It is to invest simultaneously in the AI methods that can push the boundaries of biological prediction, in the experimental biology that can validate and challenge those predictions at the level of mechanism, and in the philosophical and scientific frameworks that can sharpen the questions we ask about what understanding biological life actually requires. In doing so, we may find that the question “can AI understand life” is not a question about AI alone, but about how clearly we understand life ourselves.
References
- Hein, Z.M., Guruparan, D., Okunsai, B., et al. “AI and Machine Learning in Biology: From Genes to Proteins.” Biology (MDPI). 2025. DOI: 10.3390/biology14101453.
- Jumper, J., Evans, R., Pritzel, A., et al. “Highly accurate protein structure prediction with AlphaFold.” Nature. 2021.
- Abramson, J., Adler, J., Dunger, J., et al. “Accurate structure prediction of biomolecular interactions with AlphaFold 3.” Nature. 2024.
- Hayes, T., Rao, R., Akin, H., et al. “Simulating 500 million years of evolution with a language model.” EvolutionaryScale / Science. 2024.
- Chakraborty, S., Bhattacharya, R., and Lee, J. “The transformative impact of AI-enabled AlphaFold 3: evolution, current status, and future prospects in structural biology.” Frontiers in Artificial Intelligence. 2026.
- Zheng, H., Lin, H., Alade, A.A., et al. “AlphaFold3 in Drug Discovery: A Comprehensive Assessment of Capabilities, Limitations, and Applications.” bioRxiv. 2025. DOI: 10.1101/2025.04.07.647682.
- Passaro, S., Corso, G., and Wohlwend, J. “Boltz-2: Biomolecular Foundation Model for Joint Structure and Affinity Prediction.” MIT Jameel Clinic / Recursion Pharmaceuticals. 2025.
- Ferruz, N., and colleagues. “Explainable AI for protein language models: a call to action.” Nature Machine Intelligence. 2026.
- Noé, F., De Fabritiis, G., and Clementi, C. “Machine learning for protein folding and dynamics.” Current Opinion in Structural Biology. 2020.
- Schmid, M.B., and colleagues (Estonian Research Council). “On biological and artificial consciousness: A case for biological computationalism.” Neuroscience and Biobehavioral Reviews. 2026. DOI: 10.1016/j.neubiorev.2025.106524.
- Safron, A., Levin, M., Klimaj, V., et al. “World models, artificial general intelligence and the hard problems of life-mind continuity: toward a unified understanding of natural and artificial intelligence.” Philosophical Transactions of the Royal Society A. 2026. DOI: 10.1098/rsta.2024.0533.
- Cleeremans, A., and colleagues (Universite Libre de Bruxelles). “Explaining how consciousness emerges: current priorities and implications.” ScienceDaily. 2026.
- McClelland, T. (University of Cambridge). “What if AI becomes conscious and we never know.” ScienceDaily. 2025.
- Dehghani, N., and Levin, M. “Bio-inspired AI: Integrating Biological Complexity into Artificial Intelligence.” arXiv. 2024. arXiv:2411.15243.
- Alavi, A., Akhoundi, H., and Kouchmeshki, F. “Analyzing Advanced AI Systems Against Definitions of Life and Consciousness.” arXiv. 2025. arXiv:2502.05007.
- Bessemer Venture Partners. “Building biology-native data infrastructure for the AI era.” Bessemer Venture Partners Atlas. 2026.
- Listov, D., Goverde, C.A., Correia, B.E., and Fleishman, S.J. “Opportunities and challenges in design and optimization of protein function.” Nature Reviews Molecular Cell Biology. 2024.
- Haghighi, F., Bunne, C., Regev, A., and Cui, A. “scGPT: toward building a foundation model for single-cell multi-omics using generative AI.” Nature Methods. 2024.