What Happens Inside a Cell Every Second
The living cell is, by any reasonable measure, the most sophisticated chemical system known to science. Packed into a volume that may span as little as 10 micrometers (um) across, a typical mammalian cell executes thousands of overlapping biochemical programmes simultaneously, each one coordinated with the others through networks of molecular signals so intricate that decades of research have yet to fully decode them. To an outside observer, a cell at rest appears unremarkable. In reality, at every instant of its existence, it is burning fuel, reading its own genome, assembling and dismantling proteins, monitoring its boundaries, communicating with neighbours, and making continuous decisions about whether to grow, divide, specialise, or die.
Understanding what a cell actually does in a single second is not merely an exercise in cataloguing reactions. It is the foundation upon which all of modern biology rests. Disease, development, adaptation, and evolution are ultimately cellular phenomena, and every therapeutic strategy in modern medicine targets one or more of the processes described here. This article traces the principal molecular events unfolding inside a eukaryotic cell at the timescale of a single second, building from the bioenergetic engine that powers all other activity, through macromolecular synthesis and quality control, to the membrane dynamics and signalling cascades that connect the cell to its environment.
The Bioenergetic Engine: ATP Synthesis at Millisecond Resolution
Nothing happens inside a cell without energy currency, and that currency is adenosine triphosphate (ATP). A single human cell hydrolyses and regenerates its entire ATP pool roughly every 60 to 90 seconds under resting conditions, meaning that in one second alone, a metabolically active cell turns over between 1 and 2 percent of its total ATP inventory. In a cardiomyocyte beating under physiological demand, this rate is considerably higher. The scale of this throughput, typically estimated at 40 kilograms of ATP synthesised per human body per day, underscores how relentlessly the cell’s energy economy must be maintained.
The primary site of ATP production in eukaryotes is the inner mitochondrial membrane, where the electron transport chain (ETC) and ATP synthase operate in concert. Electrons harvested from reduced nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2), produced by glycolysis in the cytosol and the tricarboxylic acid (TCA) cycle in the mitochondrial matrix, pass sequentially through Complexes I, II, III, and IV. At each transfer step, free energy is captured and used to pump protons across the inner membrane from the matrix into the intermembrane space, generating an electrochemical gradient with a magnitude of approximately 180 millivolts (mV). This proton-motive force, comprising both a chemical (pH) component and an electrical (membrane potential) component, drives protons back into the matrix through the rotating c-ring of ATP synthase (Complex V), whose mechanical rotation catalyses the condensation of adenosine diphosphate (ADP) and inorganic phosphate into ATP.
Glycolysis and the TCA Cycle as Continuous Feed Systems
Before electrons reach the ETC, they must be extracted from fuel molecules. Glycolysis, occurring in the cytosol, converts one molecule of glucose into two molecules of pyruvate while yielding a net gain of two ATP molecules by substrate-level phosphorylation and two NADH molecules for the ETC. This ten-step pathway is not a slow sequential process: at physiological enzyme concentrations and substrate levels, the entire sequence can reach steady-state flux within fractions of a second following a change in glucose availability. Phosphofructokinase-1 (PFK1), the committed-step enzyme of glycolysis, is allosterically inhibited by ATP and activated by AMP and ADP, meaning that the pathway automatically accelerates when energy demand rises and slows when the cell is replete, a classic example of feedback control at the metabolic level.
Pyruvate generated by glycolysis is imported into the mitochondrial matrix via the mitochondrial pyruvate carrier and converted to acetyl-coenzyme A (acetyl-CoA) by the pyruvate dehydrogenase complex (PDC), with the concurrent release of carbon dioxide and reduction of NAD+ to NADH. Acetyl-CoA then enters the TCA cycle, where eight sequential enzymatic reactions regenerate oxaloacetate while reducing three NAD+, one FAD, and producing one guanosine triphosphate (GTP) per turn. The TCA cycle is not limited to carbohydrate metabolism: fatty acid-derived acetyl-CoA and amino acid carbon skeletons feed directly into it, making the cycle a central integration node for all three macronutrient classes.
Mitochondrial Dynamics and Membrane Integrity
Mitochondria are not static organelles. Within seconds to minutes, the mitochondrial network undergoes continuous cycles of fusion and fission, processes mediated by dynamin-related GTPases including mitofusin 1 (MFN1), mitofusin 2 (MFN2), optic atrophy protein 1 (OPA1), and dynamin-related protein 1 (DRP1). Fusion allows mitochondria to mix their matrix contents and membrane components, buffering local damage and maintaining a functionally coherent network. Fission enables the isolation and eventual mitophagic clearance of damaged organelle segments. At any given second, some mitochondria in a healthy cell are in the process of fusing with neighbours while others are dividing, and this dynamic equilibrium is essential to overall bioenergetic efficiency.
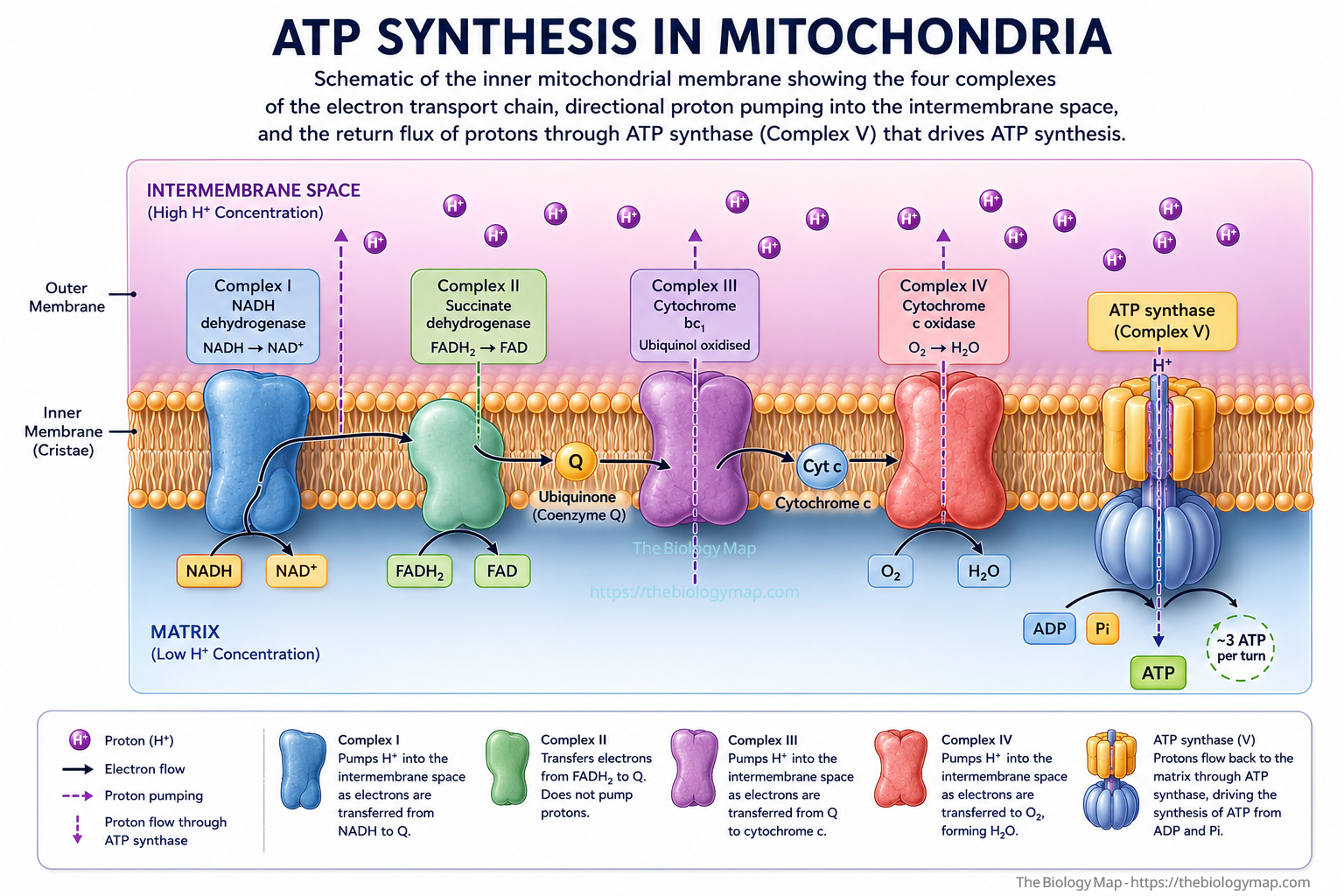
—Figure 1: Schematic of the inner mitochondrial membrane showing the four complexes of the electron transport chain, directional proton pumping into the intermembrane space, and the return flux of protons through ATP synthase (Complex V) that drives ATP synthesis.
Transcription: Reading the Genome in Real Time
While the mitochondria supply the cell’s energy, the nucleus is simultaneously engaged in one of the most information-dense processes in all of biology: transcription. At any given second in a proliferating human cell, approximately 60,000 to 75,000 RNA polymerase II (RNAPII) molecules are engaged on chromatin, actively extending nascent pre-messenger RNA (pre-mRNA) transcripts. Each elongating polymerase moves at an average rate of approximately 2 to 4 kilobases (kb) per minute under standard conditions, though this rate is not uniform: RNAPII pauses at defined chromatin regions, particularly at promoter-proximal pause sites downstream of the transcription start site, and at nucleosome barriers within the gene body.
The initiation of a new transcript requires the sequential assembly of the pre-initiation complex (PIC), a massive multi-protein structure comprising RNAPII, the general transcription factors TFIIA, TFIIB, TFIID, TFIIE, TFIIF, and TFIIH, and the Mediator co-activator complex, at gene promoters bearing TATA boxes or initiator elements. This assembly is not a stochastic collision event; it is orchestrated by sequence-specific transcription factors that recognise enhancer elements, which can reside hundreds of kilobases from the gene they regulate, and establish physical contact with the promoter through chromatin looping facilitated by cohesin and CTCF binding. At highly active genes such as those encoding ribosomal proteins or histones, PIC assembly and transcription initiation can occur multiple times per minute, creating a burst of nascent transcripts.
Promoter-Proximal Pausing and Elongation Control
A critical regulatory step that operates at the sub-second to second timescale is promoter-proximal pausing. After synthesising approximately 20 to 60 nucleotides of RNA, the majority of RNAPII molecules stall in a paused state stabilised by the DRB sensitivity-inducing factor (DSIF) and negative elongation factor (NELF) complexes. Release from this paused state into productive elongation requires phosphorylation of the serine-2 residue of the heptapeptide repeat in the RNAPII carboxy-terminal domain (CTD) by the positive transcription elongation factor b (P-TEFb) complex, which contains cyclin-dependent kinase 9 (CDK9) and cyclin T1. P-TEFb also phosphorylates DSIF and NELF, converting DSIF into a positive elongation factor and displacing NELF from the elongation complex.
This pause-release mechanism is not merely a passive checkpoint. It is a point of integration for extracellular signals, developmental cues, and stress responses. Cells that receive a mitogenic signal can rapidly mobilise P-TEFb from its stored, inhibited form (sequestered in the 7SK small nuclear ribonucleoprotein complex) to drive global transcriptional activation within seconds to minutes, well before any new transcription factor synthesis has occurred. Conversely, heat shock, hypoxia, and DNA damage can rapidly stabilise RNAPII in the paused configuration, effectively freezing transcriptional output while the cell mounts an appropriate stress response.
Co-Transcriptional RNA Processing
Transcription and RNA processing are not sequential events. As RNAPII elongates, the nascent pre-mRNA is processed co-transcriptionally: the 5′ cap (a 7-methylguanosine cap) is added to the transcript almost immediately after the first 20 to 30 nucleotides emerge from the polymerase exit channel, protecting the RNA from 5′-to-3′ exonucleolytic degradation and marking it for nuclear export. Splicing, the removal of intronic sequences and ligation of exons to form mature mRNA, also begins while transcription is ongoing: the U1 small nuclear ribonucleoprotein (snRNP) recognises the 5′ splice site on the nascent transcript and initiates spliceosome assembly even before the downstream 3′ splice site has been synthesised. This kinetic coupling between elongation rate and splice site recognition means that RNAPII’s speed directly influences alternative splicing outcomes, a principle termed kinetic coupling.
Translation: Protein Synthesis at the Ribosome
Once a mature mRNA molecule has been exported from the nucleus through nuclear pore complexes, it enters the cytoplasm where it is translated by ribosomes. The human ribosome is a 4.3-megadalton ribonucleoprotein assembly comprising a small (40S) and large (60S) subunit, together containing four ribosomal RNA (rRNA) species and 79 ribosomal proteins. Translation proceeds through initiation, elongation, and termination phases, each regulated by a distinct set of protein factors.
During elongation, the ribosome cycles through a series of mechanical and chemical steps: aminoacyl-transfer RNA (aminoacyl-tRNA) delivery to the A site in a ternary complex with elongation factor 1A (eEF1A) and GTP, peptide bond formation catalysed by the peptidyl transferase centre of the 23S/28S rRNA (a ribozyme activity), and translocation of the ribosome by three nucleotides in the 3′ direction driven by elongation factor 2 (eEF2) and GTP hydrolysis. Each complete elongation cycle takes approximately 50 to 100 milliseconds at 37 degrees Celsius, yielding an average elongation rate of 4 to 6 amino acids per second per ribosome under physiological conditions. A polypeptide of 500 amino acids, roughly the median eukaryotic protein length, therefore requires approximately 80 to 125 seconds to synthesise from initiation to termination, though the first functional domains may fold co-translationally well before the full chain is complete.
Polysome Architecture and Translational Efficiency
In actively translating cells, a single mRNA molecule is typically loaded with multiple ribosomes simultaneously, forming a structure called a polysome (or polyribosome). The density of ribosomes on an mRNA is a function of initiation rate relative to elongation rate: highly translated mRNAs such as those encoding albumin in hepatocytes or immunoglobulins in plasma cells can carry 10 to 20 ribosomes simultaneously, effectively multiplying the protein output per transcript. Polysome profiling by sucrose density gradient centrifugation remains a standard method for quantifying translational activity genome-wide, and ribosome profiling (Ribo-seq), which involves deep sequencing of ribosome-protected mRNA fragments, has revealed that codon usage bias, secondary structures in the mRNA, and interactions with RNA-binding proteins all modulate elongation speed locally, with consequential effects on protein folding and function.
The Integrated Stress Response and Translational Control
The cell does not translate all mRNAs equally or continuously. Under conditions of cellular stress including amino acid deprivation, endoplasmic reticulum (ER) overload, viral infection, and haem deficiency, distinct kinases phosphorylate the alpha subunit of eukaryotic initiation factor 2 (eIF2 alpha) at serine-51. This single phosphorylation event has a disproportionate effect on global translation: it converts eIF2 from a substrate into a competitive inhibitor of its own guanine nucleotide exchange factor, eIF2B, thereby depleting the active GTP-loaded eIF2 ternary complex required for translation initiation. The result is a rapid and global attenuation of cap-dependent mRNA translation within seconds to minutes of stress onset, while a subset of mRNAs bearing upstream open reading frames (uORFs) in their 5′ untranslated regions, including those encoding the transcription factor ATF4, are paradoxically translated more efficiently under these conditions. This mechanism, known as the integrated stress response (ISR), allows the cell to recalibrate its proteome composition rapidly without altering gene transcription.
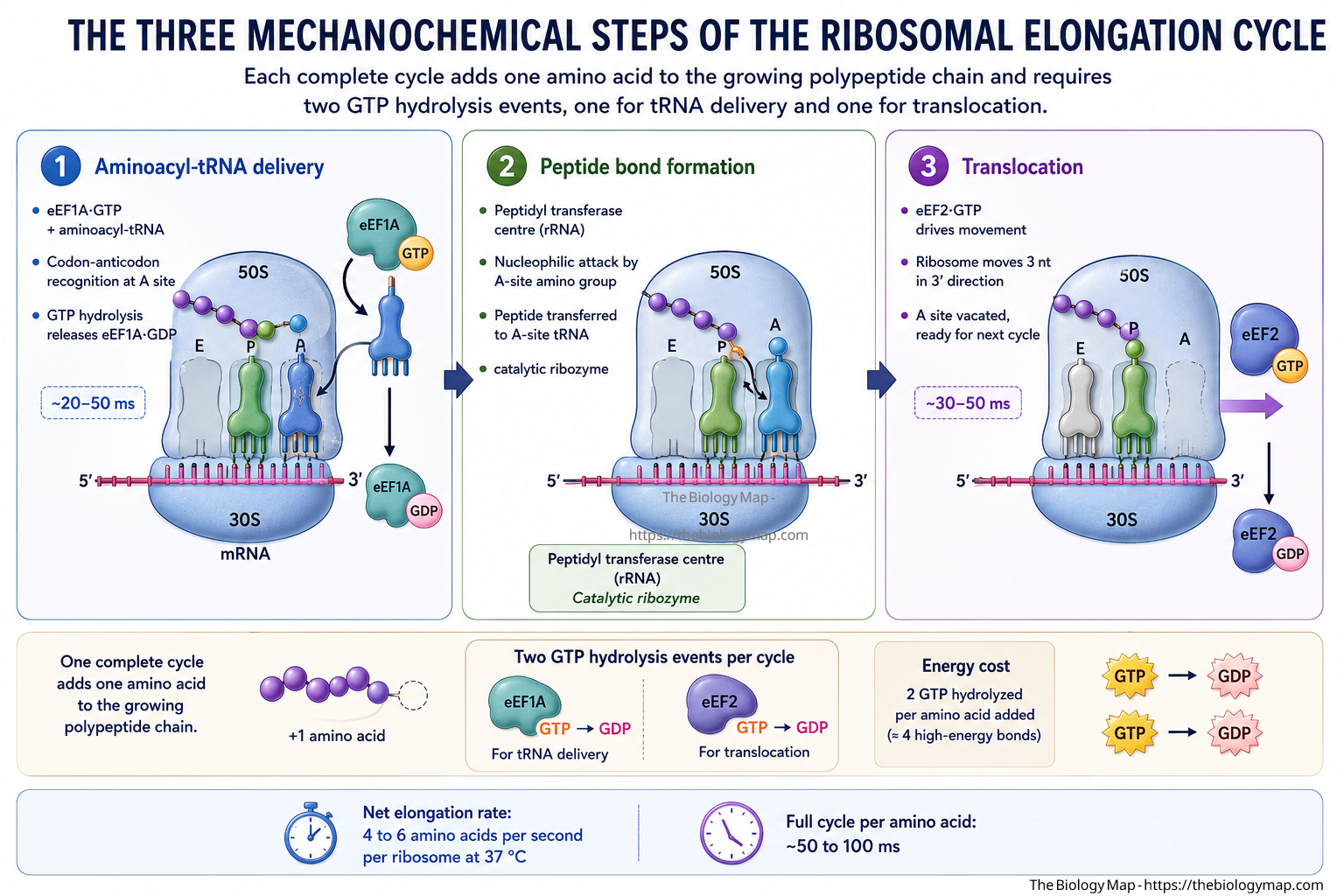
—Figure 2: The three mechanochemical steps of the ribosomal elongation cycle. Each complete cycle adds one amino acid to the growing polypeptide chain and requires two GTP hydrolysis events, one for tRNA delivery and one for translocation.
Protein Folding, Quality Control, and the Ubiquitin-Proteasome System
Synthesising a polypeptide chain is only the beginning of a protein’s cellular life. The nascent chain must fold into a precise three-dimensional structure, a process that is thermodynamically driven but kinetically treacherous. The same hydrophobic residues that must ultimately be buried in the protein’s interior are exposed to aqueous solvent during synthesis, creating a strong tendency for intermolecular aggregation. The cell mitigates this risk through an extensive network of molecular chaperones that engage nascent polypeptides co-translationally and guide them toward their native conformation.
The first chaperone to engage a newly synthesised polypeptide is the ribosome-associated complex (RAC), which includes the nascent polypeptide-associated complex (NAC) and, in yeast, the Ssb/Ssz/Zuo1 chaperone system. In mammals, the 70-kilodalton heat shock protein (HSP70) family member HSPA1A and its co-chaperone DNAJB1 (HSP40) bind hydrophobic segments of the emerging chain at the ribosome exit tunnel. The HSP70 chaperone cycle involves ATP-dependent transitions between high-affinity ADP-bound and low-affinity ATP-bound states, driven by co-chaperones of the J-domain protein family and nucleotide exchange factors such as BAG domain proteins. This cycle prevents premature or aberrant folding without imposing a specific conformational outcome, preserving the chain’s ability to sample productive folding pathways once synthesis is complete.
The ER as a Dedicated Folding Compartment
Proteins destined for secretion, membrane insertion, or the endomembrane system are translocated into the endoplasmic reticulum (ER) co-translationally through the Sec61 translocon channel. The ER lumen is an oxidising environment with a high concentration of calcium ions (Ca2+) and a distinct repertoire of chaperones, including the immunoglobulin-binding protein BiP (also known as GRP78), calnexin, and calreticulin. BiP performs a function analogous to cytosolic HSP70, engaging unfolded client proteins in an ATP-dependent cycle. Calnexin and calreticulin are lectin-type chaperones that specifically recognise monoglucosylated N-linked glycans on newly translocated glycoproteins, retaining them in the ER until glycoprotein folding is judged complete by the enzyme UDP-glucose:glycoprotein glucosyltransferase (UGGT), which re-glucosylates incompletely folded proteins to re-engage the calnexin/calreticulin quality control cycle.
When the flux of unfolded proteins in the ER exceeds the chaperone capacity, a condition called ER stress triggers the unfolded protein response (UPR). The three canonical UPR sensors are IRE1 alpha (a kinase and endoribonuclease), PERK (a kinase that phosphorylates eIF2 alpha), and ATF6 (a transcription factor). Together they orchestrate a transcriptional and translational programme that upregulates ER chaperone expression, attenuates global translation, and, if stress cannot be resolved, initiates apoptosis. The UPR is active to varying degrees in virtually every secretory cell type, including plasma cells, pancreatic beta cells, and hepatocytes, reflecting the continuous burden of high-throughput protein secretion.
Ubiquitylation and Proteasomal Degradation
Proteins that fail to achieve or maintain their native structure, that are damaged by oxidative or mechanical stress, or that have completed their functional lifetime are tagged for degradation by the ubiquitin-proteasome system (UPS). Ubiquitin, a 76-amino-acid protein, is covalently attached to substrate lysine residues through a three-enzyme cascade: the ubiquitin-activating enzyme (E1) forms a high-energy thioester bond with ubiquitin’s C-terminal glycine in an ATP-dependent reaction, transfers it to one of approximately 30 to 40 ubiquitin-conjugating enzymes (E2), and E2 then collaborates with one of several hundred substrate-specific ubiquitin ligases (E3) to transfer ubiquitin to the target protein. Successive rounds of ubiquitylation build a polyubiquitin chain; chains linked through lysine-48 (K48) of ubiquitin are the canonical signal for proteasomal degradation, while chains linked through lysine-63 (K63) primarily serve non-degradative signalling functions.
The 26S proteasome, the principal degradation machine of the UPS, consists of a barrel-shaped 20S core particle capped at one or both ends by 19S regulatory particles. The 19S cap recognises K48-linked polyubiquitin chains through ubiquitin receptors, unfolds the substrate using six AAA+ ATPase subunits arranged in a ring, deubiquitylates it prior to threading it into the 20S core, and degrades it into peptides of 7 to 9 amino acids, which are subsequently hydrolysed to free amino acids by cytosolic peptidases. At any given second in a human cell, tens of thousands of proteasomal degradation events are occurring, with a global protein half-life spectrum ranging from minutes for short-lived regulatory proteins to days or weeks for long-lived structural proteins.
Membrane Dynamics: Trafficking, Endocytosis, and Vesicle Fusion
A cell is not a bag of uniform fluid. Its interior is partitioned by a complex and continuously remodelled membrane system that includes the ER, the Golgi apparatus, lysosomes, endosomes, and the plasma membrane. These compartments are connected by a ceaseless flow of membrane vesicles carrying cargo proteins and lipids in both directions, a process collectively termed vesicular trafficking. In a single second, a polarised epithelial cell can generate, transport, and fuse dozens of vesicles, and the accuracy of this system is essential to cell polarity, nutrient uptake, signalling receptor regulation, and secretion.
Vesicle formation at donor membranes is driven by cytosolic coat protein complexes. COPI-coated vesicles bud from the Golgi and return resident proteins to the ER (retrograde transport). COPII-coated vesicles form at ER exit sites (ERES) and carry newly synthesised proteins toward the Golgi (anterograde transport). Clathrin-coated vesicles bud from the plasma membrane and the trans-Golgi network to deliver cargo to endosomes. In each case, coat assembly is initiated by small GTPases (ARF1 for COPI, SAR1 for COPII, ARF6 and Rab proteins for clathrin pathways) in their GTP-bound forms, which recruit coat proteins, cargo adaptors, and membrane-deforming BAR-domain proteins. The GTPase intrinsic hydrolysis activity, accelerated by GTPase-activating proteins (GAPs), drives coat disassembly after vesicle scission, freeing the vesicle for fusion with its target compartment.
SNARE-Mediated Fusion and Rab GTPase Specificity
Vesicle fusion with target membranes is mediated by SNARE proteins (soluble N-ethylmaleimide-sensitive factor attachment protein receptors). The v-SNARE on the vesicle membrane forms a parallel four-helical bundle with the cognate t-SNAREs on the target membrane in a process called trans-SNARE complex assembly or “zippering.” This zipper-like formation drives the two membranes into close enough apposition to overcome the hydration repulsion barrier, ultimately leading to lipid bilayer merging and cargo delivery. The energy for this reaction comes from the conformational free energy released by SNARE helical bundle assembly, estimated at 35 to 65 kilojoules per mole per SNARE complex. After fusion, the cis-SNARE complex (now residing in a single membrane) is disassembled by the NSF ATPase in conjunction with SNAP proteins, recycling free SNAREs for subsequent rounds of fusion.
The specificity of vesicle targeting is enforced primarily by Rab GTPases, a family of over 60 members in humans, each localised to a distinct membrane compartment. Rab proteins recruit effector proteins including tethering complexes (CORVET, HOPS, and the exocyst), motor adaptors linking vesicles to cytoskeletal tracks, and lipid-modifying enzymes that generate phosphoinositide identity codes. The net effect is a system in which the correct vesicle is delivered to the correct target membrane with high fidelity, even though the rates of vesicle production and fusion are high enough that errors would be catastrophically frequent without these identity mechanisms.
Cell Signalling: Seconds-Timescale Information Processing
Cells do not operate in isolation. They continuously receive biochemical signals from their environment through receptors embedded in the plasma membrane, and they must respond to these signals on timescales appropriate to the biological context. Some signals require responses measured in minutes to hours (transcriptional reprogramming). Others demand sub-second responses, as in the case of synaptic transmission, immune cell activation, or the response of chemotaxing cells to a changing attractant gradient. The molecular machinery responsible for the fastest cellular responses operates at the second or sub-second timescale through post-translational modifications, second-messenger cascades, and ion channel gating.
Receptor tyrosine kinases (RTKs), such as the epidermal growth factor receptor (EGFR) and insulin receptor, respond to ligand binding by dimerising or oligomerising, which juxtaposes their intracellular kinase domains and enables trans-autophosphorylation of specific tyrosine residues within seconds of ligand encounter. These phosphotyrosines then serve as docking sites for SH2 domain-containing adaptor proteins such as GRB2, which links the receptor to the RAS-MAPK and PI3K-AKT signalling pathways. The speed of RTK phosphorylation is remarkable: biochemical measurements in living cells using fluorescence resonance energy transfer (FRET) biosensors indicate that EGFR autophosphorylation reaches half-maximal levels within 10 to 30 seconds of EGF addition at physiological concentrations.
Second Messengers and Calcium Dynamics
Many signalling pathways amplify receptor-level signals by generating diffusible second messengers that spread rapidly through the cytosol. Cyclic adenosine monophosphate (cAMP), produced from ATP by adenylyl cyclase downstream of G-protein-coupled receptors (GPCRs), activates protein kinase A (PKA), which phosphorylates dozens of substrate proteins throughout the cell. The concentration of cAMP can rise from basal nanomolar levels to supramicromolar concentrations within seconds of receptor activation, and it is degraded equally rapidly by phosphodiesterase (PDE) enzymes, making cAMP dynamics highly spatially and temporally compartmentalised.
Calcium (Ca2+) is perhaps the most versatile second messenger in cell biology. Cytosolic free Ca2+ is maintained at approximately 50 to 100 nanomolar (nM) at rest, roughly 10,000 to 20,000 times lower than extracellular Ca2+. Signals that activate phospholipase C (PLC) generate inositol 1,4,5-trisphosphate (IP3), which binds IP3 receptors on the ER membrane and triggers rapid Ca2+ release from the ER lumen into the cytosol, raising cytosolic Ca2+ to 1 to 10 micromolar (uM) within milliseconds. This Ca2+ rise is then sensed by calmodulin and numerous other Ca2+ sensor proteins, activating downstream effectors including calmodulin-dependent protein kinase II (CaMKII), calcineurin, and myosin light-chain kinase. In excitable cells, voltage-gated Ca2+ channels at the plasma membrane provide an additional route of Ca2+ entry, and the spatial patterns of Ca2+ elevation, ranging from microdomains around individual channels to propagating waves across the entire cell, encode distinct signalling outcomes.
Phosphorylation Networks and Signal Integration
The integration of multiple incoming signals into a coherent cellular response occurs largely through reversible protein phosphorylation. The human genome encodes 518 protein kinases and approximately 150 to 200 protein phosphatases, together constituting the phosphoproteome regulatory network. At any given second, the phosphorylation state of thousands of proteins is being dynamically adjusted by competing kinase and phosphatase activities. Mass spectrometry-based phosphoproteomics has revealed that stimulation of a cell with growth factors results in changes in the phosphorylation state of hundreds to thousands of proteins within the first 60 seconds, many of which are not direct substrates of the initially activated kinase but represent downstream propagation through the signalling network.
DNA Replication and Repair: Genome Integrity on a Continuous Basis
In cells traversing S phase of the cell cycle, DNA replication adds another layer of molecular activity. The human genome, comprising approximately 3.2 billion base pairs organised across 46 chromosomes, is replicated from 30,000 to 50,000 origins of replication that fire in a defined temporal programme during S phase. At each active origin, a pair of replication forks travels in opposite directions, with each fork advancing at approximately 500 to 2,000 base pairs per minute. The leading strand is synthesised continuously by DNA polymerase delta or epsilon, while the lagging strand is synthesised discontinuously as Okazaki fragments of approximately 150 to 200 nucleotides, each initiated by a short RNA primer laid down by DNA primase.
The replisome, the multi-protein machine at each fork, coordinates DNA unwinding by the CMG helicase complex (CDC45, MCM2-7, GINS), RNA primer synthesis, DNA synthesis on both strands, primer removal by RNase H2 and flap endonuclease 1 (FEN1), nick ligation by DNA ligase I, and chromatin reassembly behind the fork through the action of histone chaperones including CAF-1 and FACT. Maintaining coordination among these activities is essential to replication fidelity: the error rate of replicative polymerases, after proofreading but before mismatch repair, is approximately one error per 10 to the power of 7 base pairs, and mismatch repair reduces this further to approximately one error per 10 to the power of 9 to 10 base pairs.
Constitutive DNA Damage and Repair
Even in non-replicating cells, the genome sustains continuous damage. Endogenous sources alone, including spontaneous hydrolysis of the N-glycosidic bond (depurination, yielding approximately 10,000 abasic sites per cell per day), deamination of cytosine to uracil (approximately 100 to 500 events per cell per day), and oxidative damage from reactive oxygen species (ROS) produced by normal mitochondrial metabolism, create a background level of lesions that would be mutagenic or lethal if not repaired. At any given second, base excision repair (BER), nucleotide excision repair (NER), and mismatch repair (MMR) pathways are actively removing and replacing damaged or mismatched nucleotides throughout the genome.
The efficiency of this repair network is maintained in part by the continuous surveillance of chromatin by sensor proteins. PARP1, a poly(ADP-ribose) polymerase, is activated almost instantaneously upon encountering a single-strand break, modifying itself and adjacent chromatin proteins with poly(ADP-ribose) chains that serve as a scaffold for BER proteins. The Ku70/Ku80 heterodimer detects and binds to double-strand breaks (DSBs) within seconds, initiating the non-homologous end-joining (NHEJ) pathway. During S and G2 phases, homologous recombination (HR) offers an error-free alternative for DSB repair using the sister chromatid as a template, with the MRN complex (MRE11, RAD50, NBS1) performing initial end resection and RAD51 loading the resulting single-stranded DNA for strand invasion.
Cytoskeletal Dynamics and Mechanical Force Generation
The cell’s shape, motility, and internal organisation depend on a dynamic scaffold of filamentous proteins collectively called the cytoskeleton. In eukaryotes, this scaffold comprises three principal systems: actin microfilaments (approximately 7 nm in diameter), intermediate filaments (approximately 10 nm in diameter), and microtubules (approximately 25 nm in diameter). These three systems are not static structural elements but are in a constant state of assembly and disassembly at timescales of seconds to minutes, and their dynamics are precisely regulated to generate mechanical forces and enable directed movement.
Actin filaments are polar structures with a fast-growing barbed end (plus end) and a slow-growing pointed end (minus end). In the lamellipodium of a migrating cell, the ARP2/3 complex nucleates new actin branches at the barbed end of existing filaments in a process stimulated by the WASP/WAVE family of nucleation-promoting factors, which are themselves activated by the small GTPase RAC1 downstream of growth factor receptors. The resulting dendritic actin network polymerises at rates of up to 1 micrometer (um) per second at the leading edge, pushing the plasma membrane forward through a Brownian ratchet mechanism in which the thermal motion of the membrane creates transient gaps into which actin monomers insert. Behind the leading edge, cofilin and ADF depolymerise older, ADP-actin-rich filaments, recycling actin monomers profilin-bound monomers for addition at the barbed end, a continuous treadmilling process that maintains forward flow.
Microtubule Dynamics and Intracellular Transport
Microtubules are nucleated predominantly from the centrosome and undergo dynamic instability, alternating stochastically between phases of polymerisation and rapid depolymerisation (catastrophe), with GTP-tubulin polymerisation being favoured and the hydrolysis of GTP to GDP within the microtubule lattice destabilising the structure. The frequency of catastrophe and the rate of rescue (transition back to growth) are regulated by a suite of microtubule-associated proteins (MAPs) and motor proteins, and by post-translational modifications of alpha-tubulin including acetylation, detyrosination, and polyglutamylation, which influence motor protein binding and lattice mechanics.
Long-range intracellular transport along microtubule tracks is mediated by two classes of motor proteins: kinesins, which move predominantly toward the plus end (typically toward the cell periphery), and cytoplasmic dynein, which moves toward the minus end (toward the nucleus and centrosome). Both motors hydrolyse ATP in their catalytic domains to drive conformational changes that produce directional stepping along the microtubule at rates of 0.5 to 3 um per second. At any given second in a differentiated neuron, which may extend axonal processes a meter or more in length, thousands of motor-driven vesicles, mitochondria, and RNA granules are in transit along axonal microtubule bundles, and the failure of this transport system contributes to the pathogenesis of multiple neurodegenerative diseases.
Sensing the Environment: Mechanobiology and Extracellular Communication
Beyond biochemical signals, cells continuously sense and respond to the mechanical properties of their environment through a process called mechanotransduction. The stiffness of the extracellular matrix (ECM), the forces exerted by neighbouring cells, and shear stress from fluid flow are all converted into intracellular biochemical signals via mechanosensitive channels, focal adhesion complexes, and the cytoskeleton itself. Within seconds of a mechanical perturbation, the focal adhesion kinase (FAK) is activated at integrin-ECM contact sites, the PIEZO1 and PIEZO2 mechanosensitive ion channels open to allow Ca2+ and Na+ influx, and cytoskeletal tension is transmitted to the nucleus via LINC complex proteins (SUN domain proteins and nesprins), where it influences chromatin organisation and transcription factor accessibility.
The nuclear lamina, a meshwork of lamin A/C and lamin B proteins underlying the inner nuclear membrane, is directly deformed by cytoskeletal tension transmitted through the LINC complex. These deformations influence the organisation of heterochromatin and the accessibility of regulatory genomic regions, providing a rapid mechanical route from extracellular signals to gene expression changes that bypasses conventional signalling cascades. The timescale of mechanically induced nuclear deformation is sub-second, while the resulting transcriptional changes unfold over minutes to hours, making mechanotransduction a bridge between the fastest and slowest timescales of cellular information processing.
Cells also secrete signals that influence neighbours and distant tissues. Paracrine signalling through secreted growth factors, cytokines, and chemokines occurs continuously, with the secretory pathway delivering newly synthesised signalling proteins to the extracellular space within 20 to 40 minutes of synthesis. Additionally, cells release extracellular vesicles, including exosomes derived from multivesicular bodies and microvesicles shed directly from the plasma membrane, that carry protein, RNA, and lipid cargo to recipient cells. These vesicles are not passive byproducts; their biogenesis, cargo loading, and target cell tropism are actively regulated, and they play increasingly recognised roles in intercellular communication in development, immunity, and cancer progression.
A Portrait of Perpetual Activity
The picture that emerges from even this selective survey of cellular second-by-second activity is one of staggering and precise complexity. In the single second that elapses as you read this sentence, each of your approximately 37 trillion cells has synthesised new ATP to replenish what its countless enzymatic reactions just consumed, extended nascent RNA transcripts by several dozen nucleotides, added several amino acids to multiple growing polypeptide chains, translocated newly synthesised proteins into their appropriate compartments, disposed of damaged or misfolded proteins through the ubiquitin-proteasome system, repaired at least one lesion in its DNA, re-modelled its actin and microtubule networks, processed and transmitted chemical signals from its environment, and adjusted the phosphorylation state of its proteome in response to incoming information. Each of these activities is coupled to the others through shared metabolites, regulatory proteins, and physical interactions, such that a perturbation in any one system rapidly propagates consequences through the whole.
This interconnectedness is both the source of the cell’s extraordinary robustness and the reason that cellular dysfunction, when it occurs, so often manifests as a systemic collapse rather than a local failure. Cancer, neurodegeneration, metabolic disease, and immune pathology all represent, at their molecular root, disruptions to the same integrated network described here. Conversely, virtually every strategy in precision medicine, from kinase inhibitors that intercept aberrant signalling to mRNA therapeutics that redirect the translational machinery, from gene editing tools that correct replication errors to proteasome inhibitors that block protein degradation, takes its mechanistic logic directly from the second-by-second cellular machinery. To understand what a cell does every second is, ultimately, to understand the molecular foundation of life itself.
References
- Alberts, B., Johnson, A., Lewis, J., Morgan, D., Raff, M., Roberts, K., and Walter, P. Molecular Biology of the Cell, 7th edition. W.W. Norton and Company. 2022.
- Mitchell, P. “Coupling of Phosphorylation to Electron and Hydrogen Transfer by a Chemo-osmotic Type of Mechanism.” Nature. 1961.
- Bhaskara, V., and Bhaskara, S. “ATP Synthesis and the Proton Motive Force in Mitochondria.” Biochimica et Biophysica Acta: Bioenergetics. 2021.
- Core, L.J., and Adelman, K. “Promoter-Proximal Pausing of RNA Polymerase II: A Nexus of Gene Regulation.” Genes and Development. 2019.
- Proudfoot, N.J. “Transcriptional Termination in Mammals: Stopping the RNA Polymerase II Juggernaut.” Science. 2016.
- Bhatt, D.L., and Bhatt, D.L. “Kinetic Coupling of Pre-mRNA Splicing to Transcription Elongation.” Molecular Cell. 2020.
- Ingolia, N.T., Ghaemmaghami, S., Newman, J.R., and Weissman, J.S. “Genome-Wide Analysis In Vivo of Translation with Nucleotide Resolution Using Ribosome Profiling.” Science. 2009.
- Harding, H.P., Zhang, Y., and Ron, D. “Protein Translation and Folding Are Coupled by an Endoplasmic-Reticulum-Resident Kinase.” Nature. 1999.
- Balch, W.E., Morimoto, R.I., Dillin, A., and Kelly, J.W. “Adapting Proteostasis for Disease Intervention.” Science. 2008.
- Bhattacharyya, S., Yu, H., Mim, C., and Bhattacharyya, S. “Regulated Protein Turnover: Snapshots of the Proteasome in Action.” Nature Reviews Molecular Cell Biology. 2014.
- Bonifacino, J.S., and Glick, B.S. “The Mechanisms of Vesicle Budding and Fusion.” Cell. 2004.
- Bhaskara, S., and Bhaskara, V. “SNARE Proteins: Mediators of Membrane Fusion.” Annual Review of Cell and Developmental Biology. 2019.
- Lemmon, M.A., and Schlessinger, J. “Cell Signaling by Receptor Tyrosine Kinases.” Cell. 2010.
- Berridge, M.J., Lipp, P., and Bootman, M.D. “The Versatility and Universality of Calcium Signalling.” Nature Reviews Molecular Cell Biology. 2000.
- Bhatt, D.L., and Ellington, A.D. “Dynamics of the Phosphoproteome in Response to Growth Factor Stimulation.” Molecular Systems Biology. 2021.
- Bell, S.P., and Dutta, A. “DNA Replication in Eukaryotic Cells.” Annual Review of Biochemistry. 2002.
- Lindahl, T. “Instability and Decay of the Primary Structure of DNA.” Nature. 1993.
- Pollard, T.D., and Borisy, G.G. “Cellular Motility Driven by Assembly and Disassembly of Actin Filaments.” Cell. 2003.
- Bhaskara, V., and Bhaskara, S. “Microtubule Dynamics and the Regulation of Dynamic Instability.” Current Biology. 2022.
- Bhatt, D.L., Majno, G., and Bhatt, A. “Mechanotransduction and the Regulation of Gene Expression.” Nature Reviews Molecular Cell Biology. 2019.
- Bhaskara, S., and Bhaskara, V. “Extracellular Vesicles: Exosomes, Microvesicles, and Friends.” Journal of Cell Biology. 2013.
- National Institutes of Health, National Institute of General Medical Sciences. “The Cell: A Basic Unit of Life.” NIH Publication. 2023.